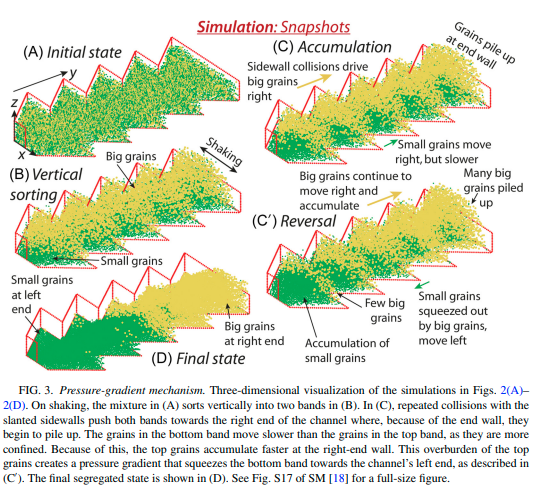
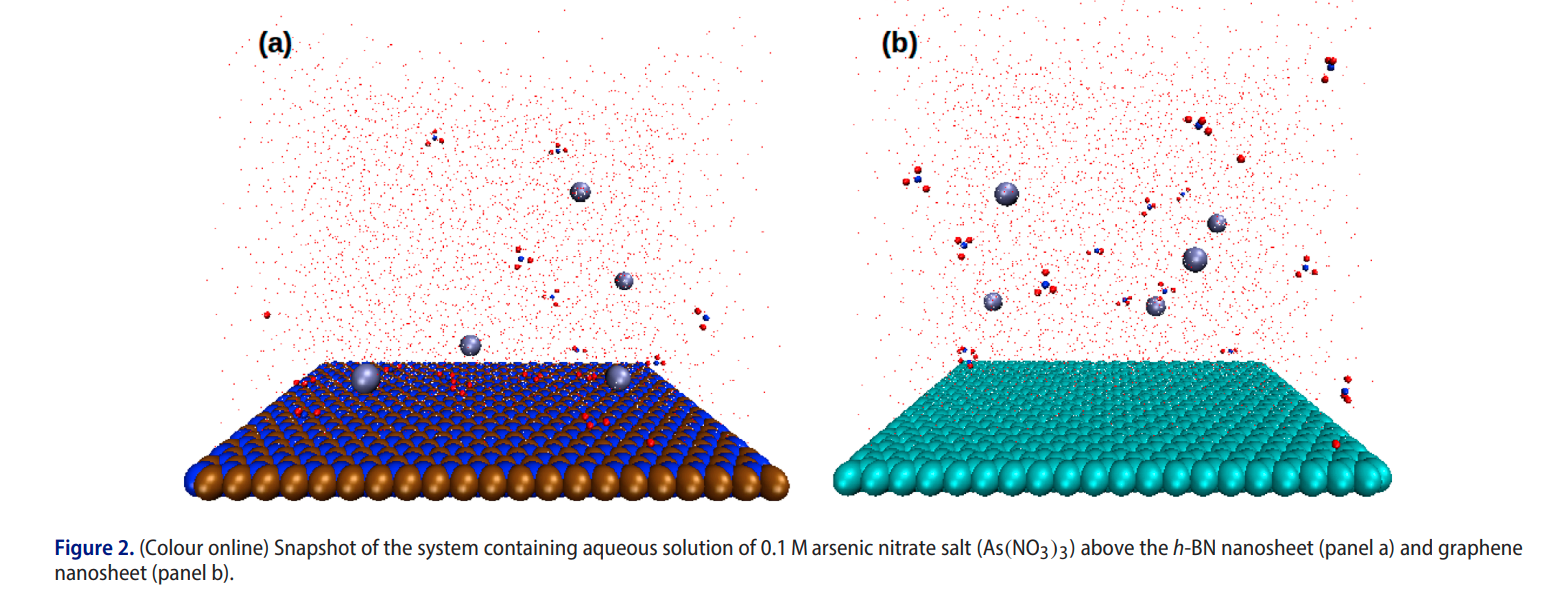
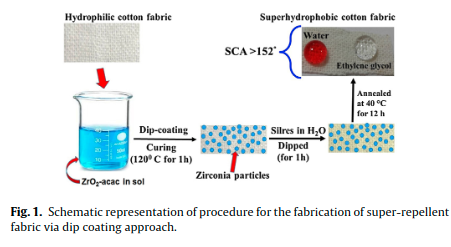
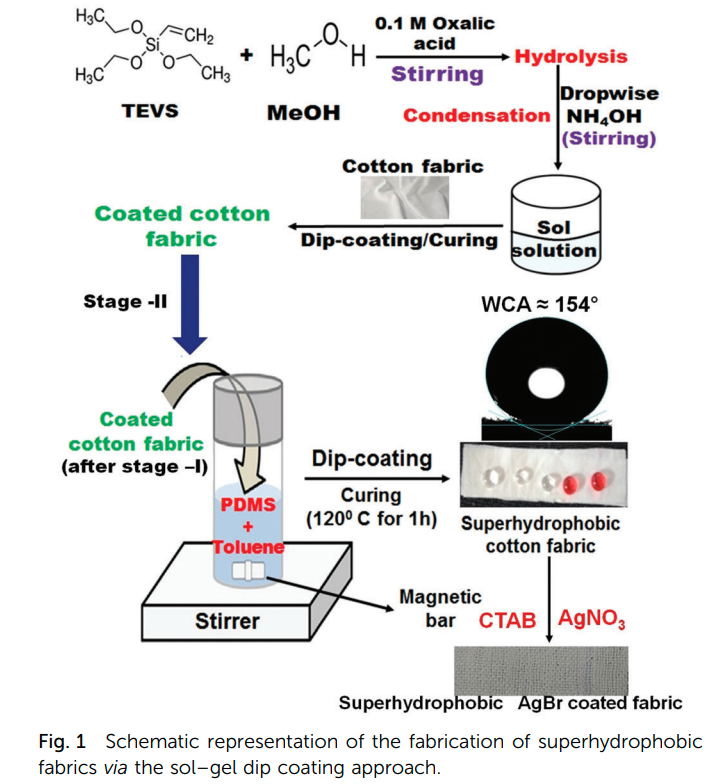
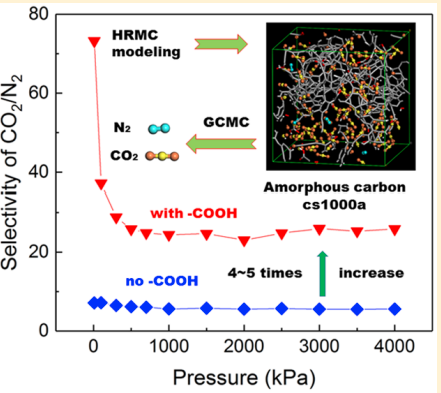
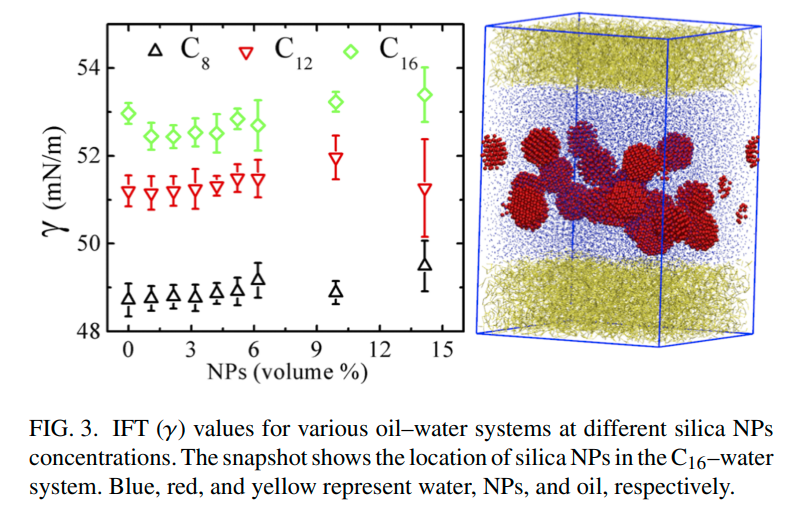
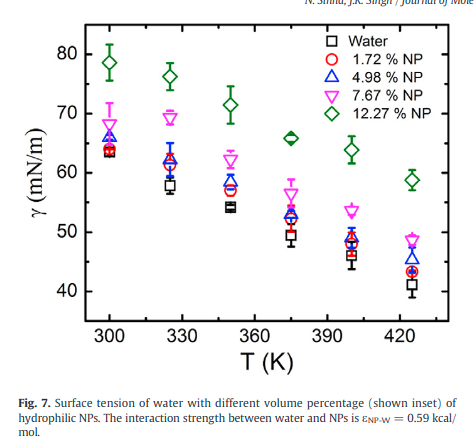
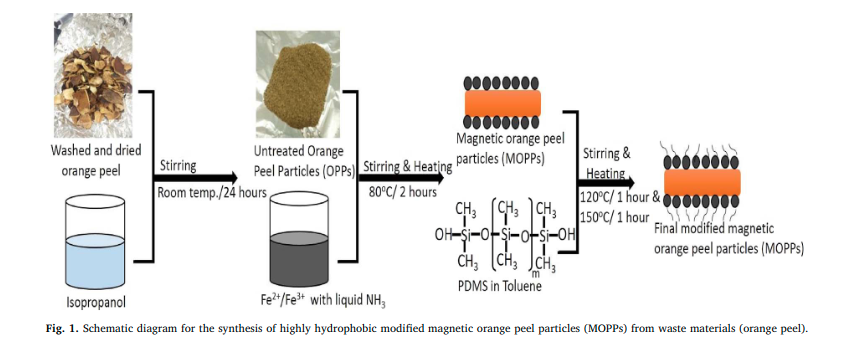
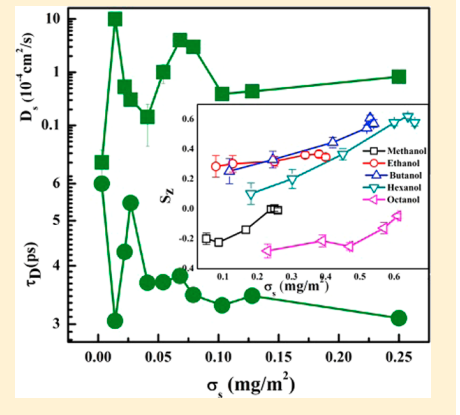
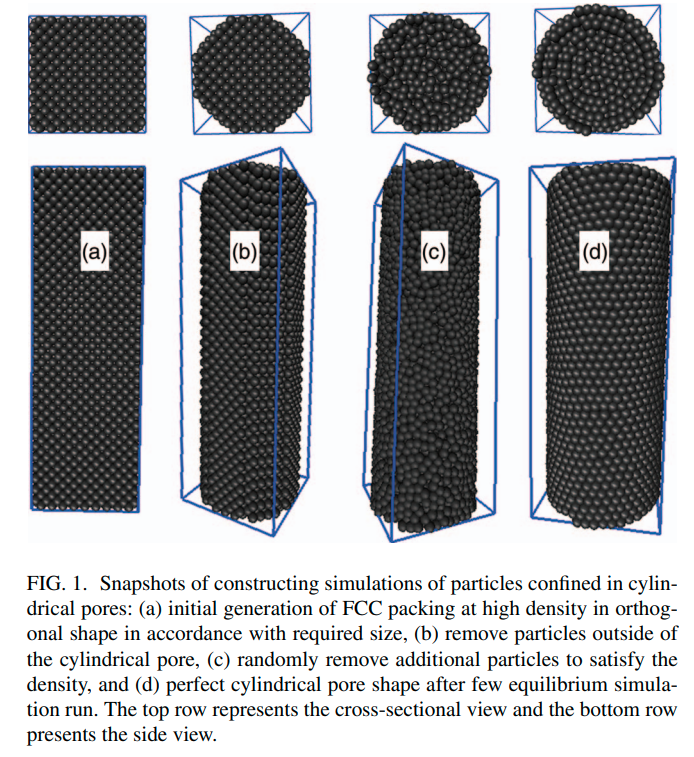
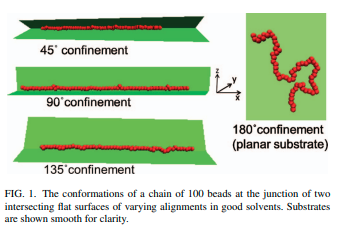
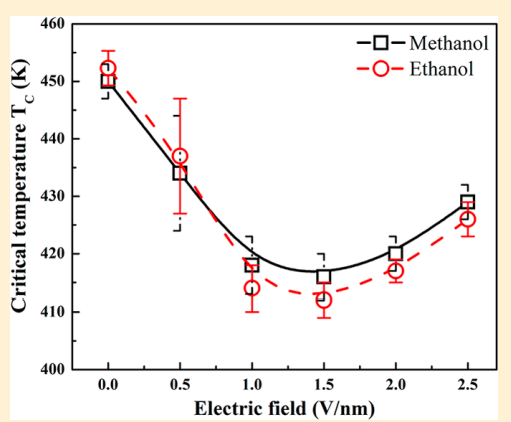
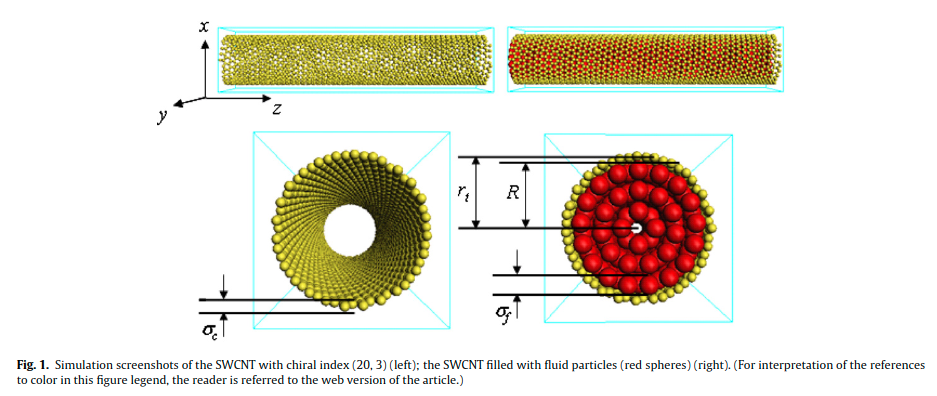
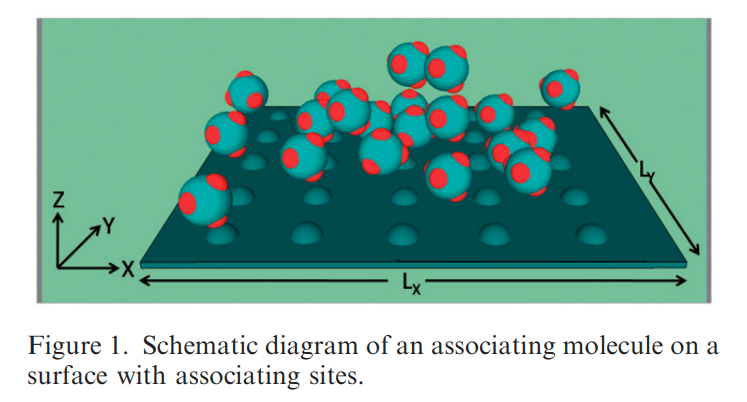
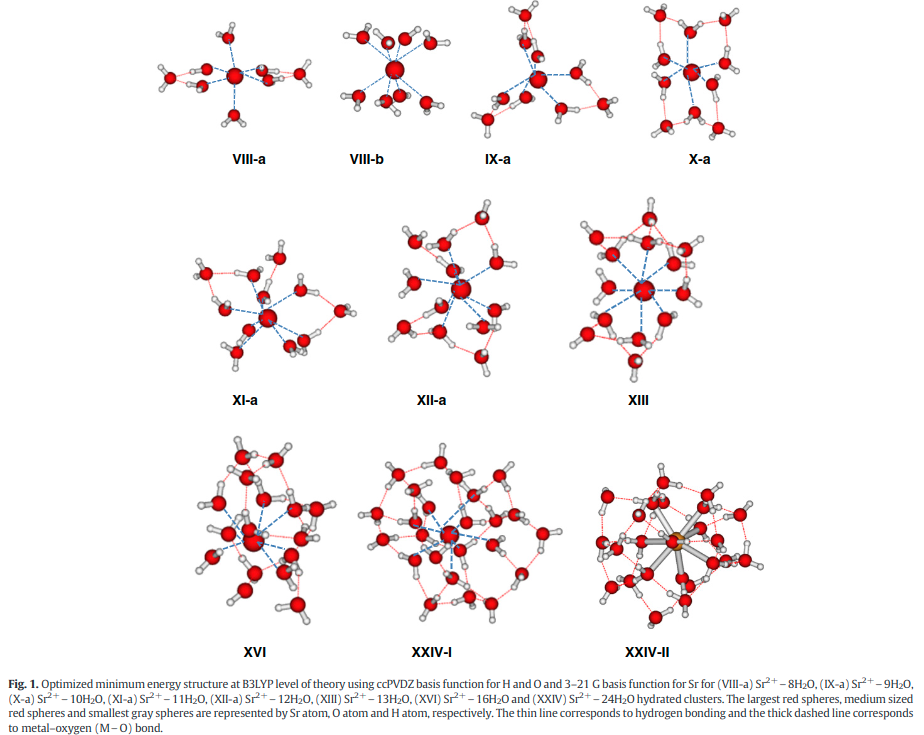
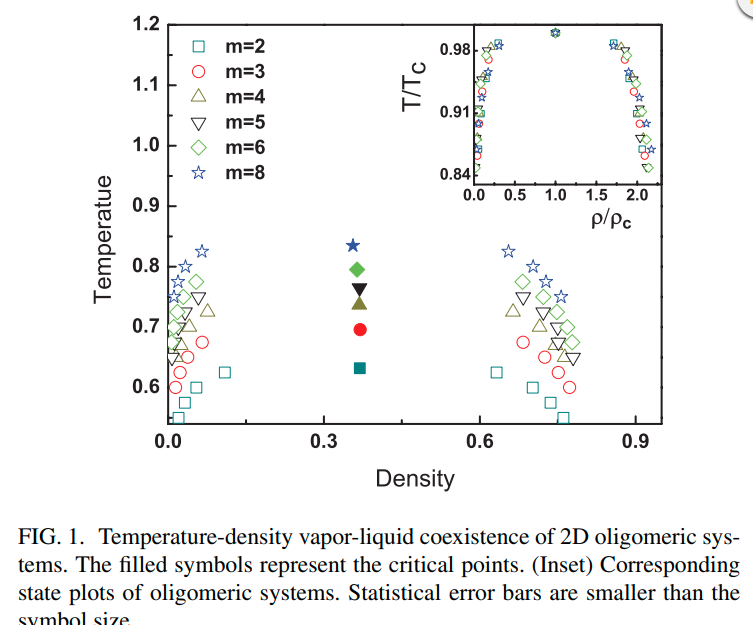
|
2026
|
|---|
-
Riya Sharma, Tamaghna Chakraborti, Anand Narayanan Krishnamoorthy, Rakesh Chandran Puthenkalathil, Harshal Chaudhari, Kuldeep Mamtani, Jayant K Singh “Does Conductivity in PiperION Depend on Molecular Size or Intermolecular Forces? Chloride versus Hydroxide Transport” ACS Applied Polymer Materials
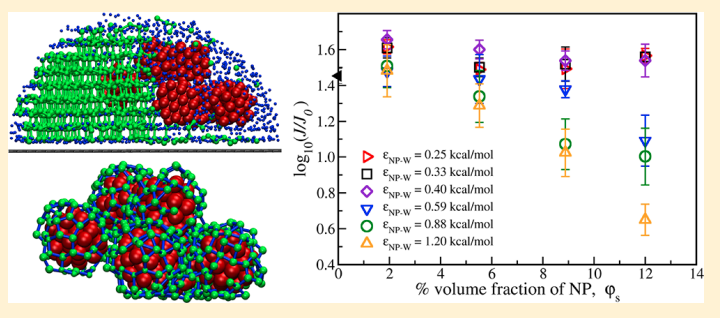
Abstract: The effect of molecular size and intermolecular interactions on the transport properties of the chloride and hydroxide ions in a PiperION anion exchange membrane is discerned using molecular dynamics simulations and a subsequent detailed comparison of the molecular features like radial distribution function (RDF), coordination number (CN), and mean square displacement (MSD). The pore characteristics of the membrane like pore limiting diameter (PLD), largest cavity diameter (LCD), and pore size distribution in the presence of the two anions are also analyzed to determine the effect of the molecular size on ionic movement through the membrane. Our results show that the anion–water RDF peak values for the hydroxide anions are more than two times that of chloride ions at all levels of hydration, implying much stronger interactions between hydroxide and water in comparison to those between chloride and water. The pore characteristics are also observed to be less in the hydroxide membrane compared with the chloride one at higher hydration levels. The stronger interactions and smaller pore sizes are observed to reduce the vehicular diffusivities of the hydroxide anion by a factor of 2.0 as compared to its chloride counterpart at high hydration levels (lambda = 8.0, 12.0, 16.0, and 20.0) when the anions are fully hydrated and remain in the aqueous phase. At low hydration levels (lambda = 4.0), however, when the pore sizes in the two samples are compatible and water channels segregated due to nonpercolation, the hydroxide ions diffuse faster on account of their smaller size compared to chloride, as is normally the case in anionic diffusion. Our simulation results are observed to match closely with experiments and other simulation data from the literature, thus verifying our findings.
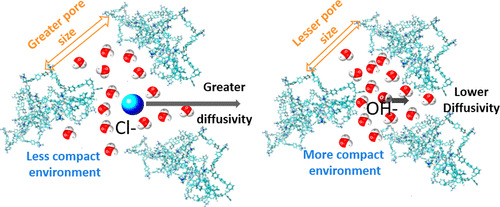
Read More ...
|
-
S Muthu Krishnan, Mona Vishwakarma, Amol Tagad, Jayant K Singh “Unravelling Ice Growth Inhibition by UiO-66 and Its Functionalized Nanoparticles: The Role of Uncoordinated Linkers in Enhancing Interfacial Interactions with Ice” ACS Applied Materials & Interfaces
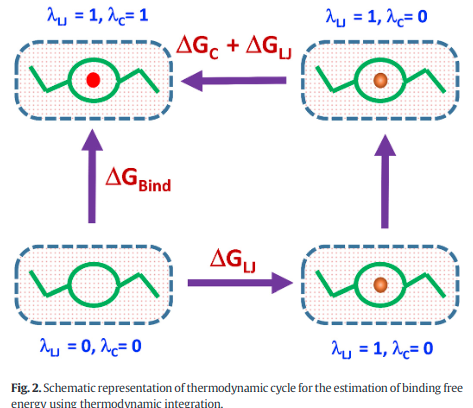
Abstract: In organ transplantation, cryogenic storage of live cells is essential, but ice recrystallization during thawing causes cell damage, necessitating effective control strategies. Ice recrystallization inhibition (IRI) agents suppress ice growth through the Gibbs–Thomson effect by binding to the ice surface. Metal–organic framework (MOF) nanoparticles, with high surface area and tunable chemistry, show strong promise as effective IRI agents; however, their inhibition mechanisms and the role of surface modifications remain insufficiently understood. In this work, we performed the first computational investigation of using UiO-66 and its functionalized derivative MOF nanoparticles with surface-modified uncoordinated linkers as IRI agents through MD simulations. We observed that the MOF nanoparticles were highly effective in inhibiting ice growth through the Gibbs–Thomson effect, with the UiO-66 nanoparticle reducing the growth rate by up to 24% compared to the benchmark ice–water system. Further, we observed that the UiO-66 nanoparticle produced the highest thermal hysteresis close to 12 K. Additionally, we showed that the adsorbed surface area of the nanoparticle plays a major role in inhibiting ice growth compared to the number of hydrogen bonds formed, deviating from previous conclusions in the literature. Through PMF calculations, we also observed that the presence of uncoordinated linkers on the MOF nanoparticle improved the binding strength of the nanoparticle to the ice surface, with UiO-66 showing a potential of mean force (PMF) of 2.9 kcal mol–1 per linker. To the best of our knowledge, this work represents the first computational investigation of ice recrystallization inhibition in MOF nanoparticles with uncoordinated linker modifications, and it establishes surface functionalization as an effective strategy for enhancing their IRI activity.
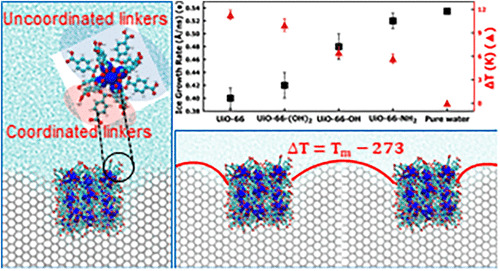
Read More ...
|
-
Diksha Srivastava, Jyotirban Dey, Soumyajit Rajak, Showkat H Mir, Jayant K Singh, Manabendra Chandra, Thiruvancheril G Gopakumar “Doping as a Strategy for Modulating Crystallinity and Optical Properties of Large Area Films of Semiconducting 2D-COFs” ACS Applied Materials & Interfaces
Abstract: Two-dimensional covalent organic frameworks (2D-COFs) represent a promising class of 2D materials with semiconducting properties, making them ideal candidates for optoelectronic applications. The key to their applicability lies in the ability to fine-tune their electronic properties and the large crystalline order in a film-like morphology. In this article, we demonstrate that the use of a dopant molecule, melamine (Me), can drastically enhance the crystallinity and modulate the optical band gap of large self-standing thin films of 2D-COF, synthesized through interfacial Schiff base condensation. 2D-COFs are synthesized using a variable ratio of p-phenylenediamine (PDA), benzene-1,3,5-tricarboxaldehyde (TCA), and melamine (Me) dopant, leading to significant alterations in both crystallinity and optical properties. Notably, the optical band gap shows a strong blue shift from 2.2 eV for the undoped 2D-COF to 2.6 eV for the optimally Me-doped 2D-COF. In addition, for the optimum ratio of melamine to PDA and TCA, we observed large, highly crystalline domains within the film. The effect of melamine doping on the structural and electronic modifications of the films was characterized using various spectroscopic and microscopic techniques and density functional theory calculations.
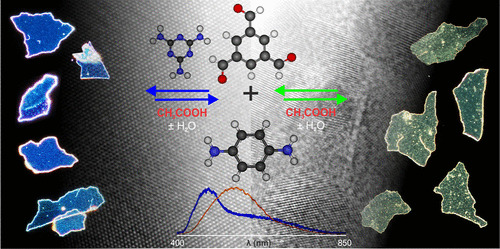
Read More ...
|
-
MV Jyothirmai, Shrish Nath Upadhyay, S Muthu Krishnan, Sudipta Roy, Jayant K Singh “From density functional theory to machine learning: Emerging paradigms in energy materials discovery” Journal of Power Sources
Abstract: Sustainable and efficient energy conversion technologies are driving significant progress in electrocatalysis, which is a crucial pathway for developing selective and high-performance catalytic materials. Density functional theory (DFT) provides a fundamental framework for understanding catalytic activity by offering quantitative insights into adsorption energetics, reaction pathways, and electronic structure-property relationships at the atomic scale. The review highlights atomic-level design strategies such as doping, defect engineering, heterostructure formation, and single-atom catalysis, which have introduced new pathways for enhancing catalytic performance by modulating active site geometry and electronic structure. We discuss dynamic simulation techniques such as ab initio molecular dynamics (AIMD), kinetic Monte Carlo (KMC), and machine learning potentials (MLP), which enhance the ability to incorporate finite-temperature effects, solvent dynamics, and reaction timescales into catalytic studies. In parallel, ML frameworks have emerged as powerful tools for high-throughput screening, property prediction, and inverse design, utilizing high-quality datasets and advanced descriptors to efficiently identify promising candidates. Through the combination of electronic structure theory and ML tools, we establish an integrated pipeline for identifying catalytic materials with improved activity and durability. We also highlight future directions, including generative models for inverse design, active learning to enable autonomous discovery, multiscale simulation strategies, and the development of physics-informed neural networks that embed domain-specific knowledge.
Read More ...
|
|
|
|
2025
|
|---|
|
|
-
Sikandar Bind, Jayant K Singh, Himanshu Sharma “Effect of brine composition on the surface energy of calcite and adhesion of oil: Implications for the wettability of limestone rocks” Journal of Molecular Liquids
Abstract: Limestone rocks hold a substantial portion of the world's existing oil reserves. However, the oil recovery from these rocks during waterflooding is low due to their oil-wet nature. The low salinity wettability alteration technique is promising in improving oil recovery from these rocks. This technique is low-cost and environmentally friendly as no chemicals, such as surfactants and polymers, are added to the injection solution. Furthermore, many limestone reservoirs exist at high temperatures and salinity conditions where these chemicals and nanoparticles are unstable. Previous studies have reported wettability alteration and additional oil recovery when appropriately designing the injection brine composition. However, the influence of brine composition on the surface energy of limestone rocks and oil adhesion is not reported in the literature. In this study, experiments and theoretical calculations were carried out to investigate the effect of brine composition on the surface energy of calcite, a dominant mineral present in limestone rocks, using the Van Oss-Chaudhury-Good (vOCG) theory. The effect of surface roughness of calcite, obtained via atomic force microscopy, on the surface energy and work of adhesion was incorporated. The surface energy was found to be 106 mJ/m2, 134 mJ/m2 and 1055 mJ/m2 for calcite surfaces aged initially in formation brine, seawater, and sulfate-rich seawater, respectively. These results show that the surface energy of calcite is lower on aging initially in the formation brine and seawater compared to sulfate-rich seawater. These results suggests that the low surface energy of limestone rocks, containing the formation brine, promotes the wetting of rock by crude oil. The work of adhesion of oil was found to be positive for calcite surfaces aged initially in the formation brine and seawater, and negative for the calcite surface aged initially in sulfate-rich seawater. These results show that the oil has a tendency to attach to calcite chips aged initially in the formation brine and seawater. Similarly, these results indicate that oil does not show adhesion towards calcite chips aged initially in sulfate-rich seawater. Overall, this study provides new insights into the wetting behavior of calcite, and presents a fundamental approach to screening promising brine compositions for increasing oil recovery from oil-wet limestone rocks.
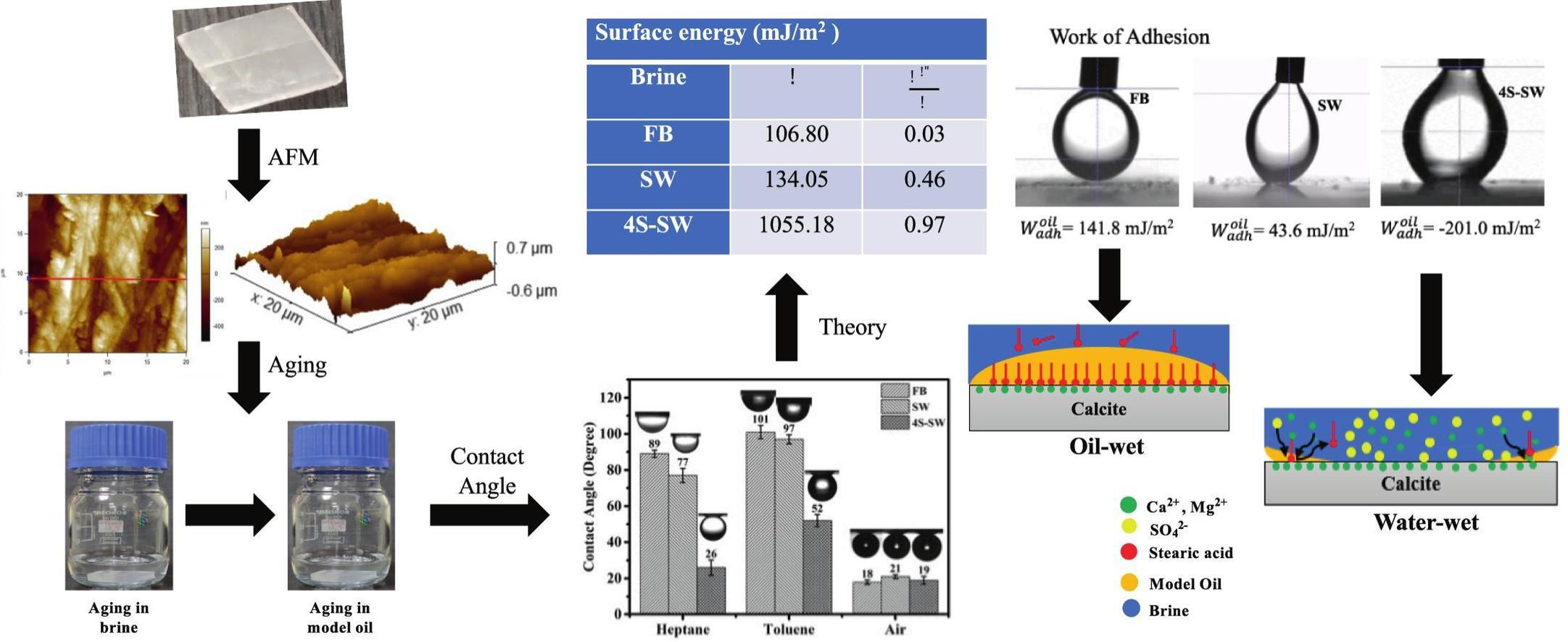
Read More ...
|
-
Riya Sharma, Tamaghna Chakraborti, Jayant K Singh “Understanding the role of alkyl chain architecture and functional group chemistry in governing ion transport in PEEK based anion exchange membranes: A molecular dynamics study” Journal of Membrane Science
Abstract: Poly(ether ether ketone) (PEEK)-based anion exchange membrane (AEM) is a promising material for green hydrogen production via water electrolysis due to its excellent mechanical, thermal, and chemical stability. This study explores the effects of alkyl side-chain length and functional group chemistry at different hydration levels on ion transport properties in PEEK based membranes. Key findings on PEEK-TMA reveal that hydration level significantly influences membrane density, ionic conductivity, and diffusivity. Density initially increases due to solvation-shell formation, then decreases as swelling occurs. Compared with experiments, vehicular diffusion alone describes chloride transport, while proton-hopping contributes roughly 60 of hydroxide conductivity. The results also reveal that the cationic group position along the alkyl chain length plays a crucial role in modulating ion transport as placing the cation at the terminal end of a longer alkyl chain length improves the mobility of the cationic site, reducing anion residence time and facilitating up to 50 faster anion diffusion without significantly altering hydrophilic pathway morphology at a fixed hydration level.
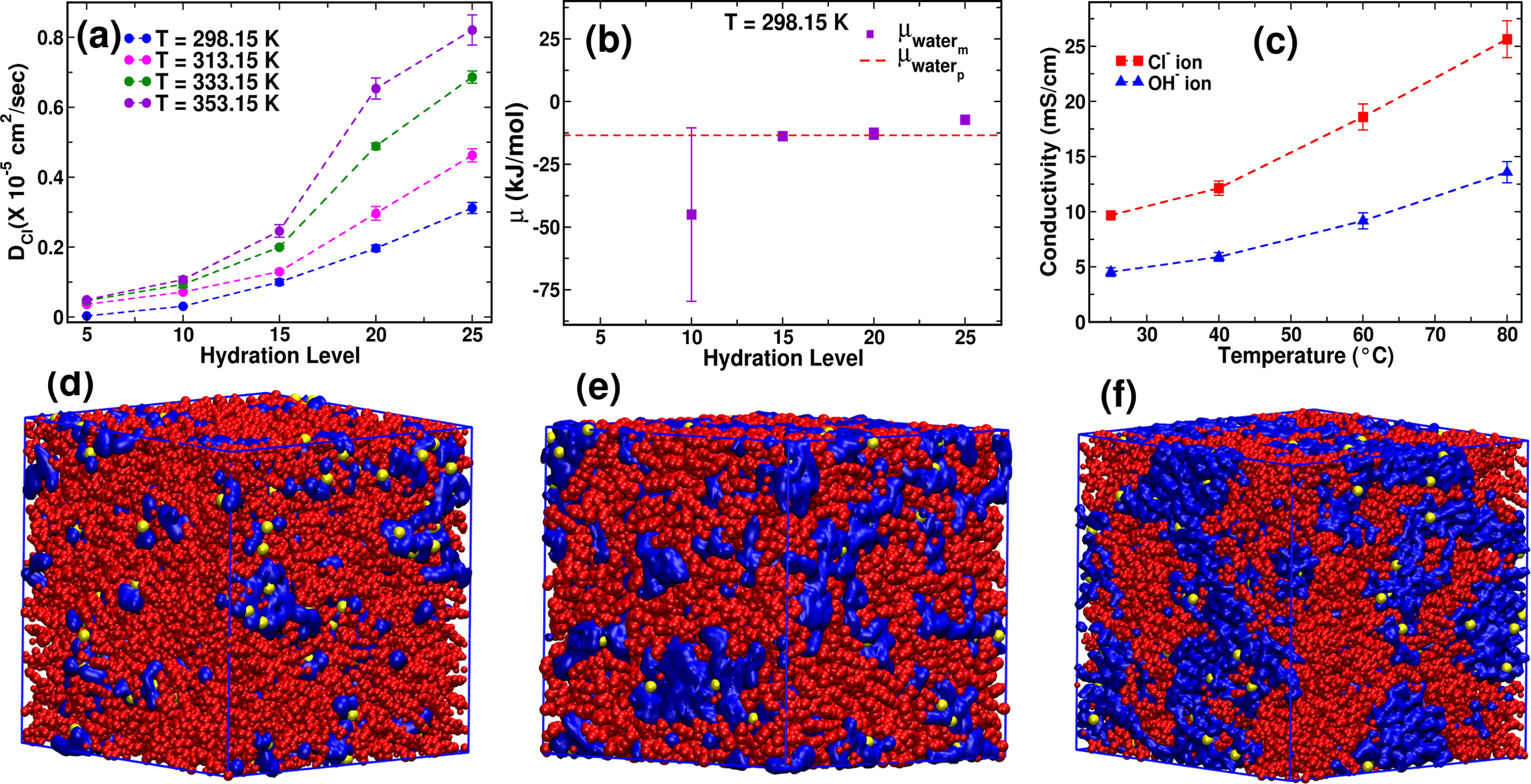
Read More ...
|
|
|
|
|
-
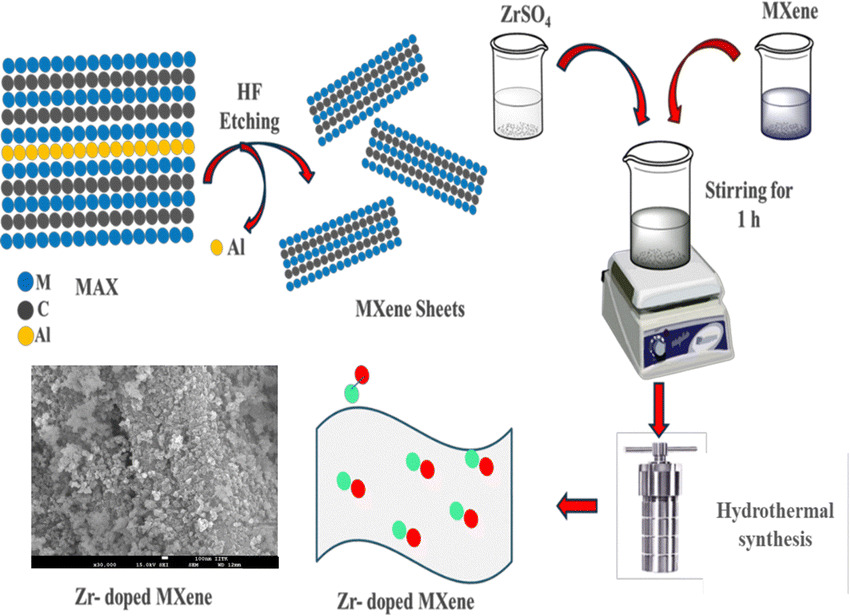
Deeksha Jaiswal, Chandan Sharma, Sudipta Roy, Prabhakar K Pandey, Abdul Quiyoom, Vinod Kumar, Pranab K Rakshit, Seetaram Chebrolu, Jayant K Singh, Jitendra K Bera “Ti3 C2 TX-based Zr@MXene for CO2 capture and conversion” Chemical Communications
Abstract: A Zr-doped MXene (Ti3C2TX) has been developed for CO2 utilization. The Zr doping enhances surface area, CO2 uptake, and active site density. The material exhibited 99% yield for styrene carbonate and significant yields with other epoxides as well. DFT calculations confirm an accessible pathway, supporting energy-efficient CO2 utilization.
Read More ...
|
-
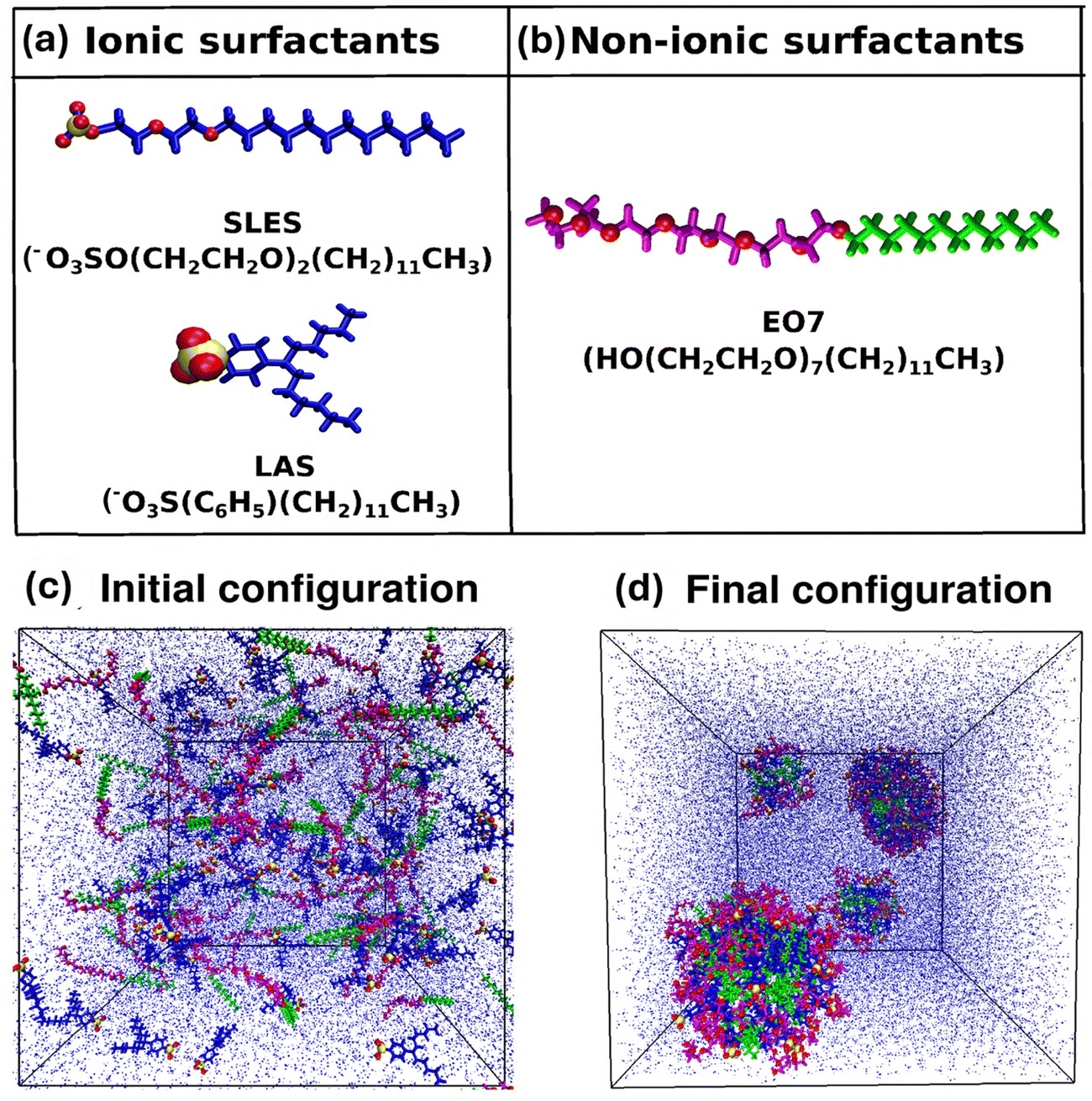
Riya Sharma, Mangesh Bhendale, Somnath Das, Samiran Mahapatra, Jayant K Singh “Morphological and microstructural insights into mixed surfactant systems: a molecular dynamics study” Soft Matter
Abstract: Surfactants are the backbone of detergent formulations, playing a crucial role in determining the cleaning performance, product aesthetics and formulation stability. The strategic combination of ionic and non-ionic surfactants has been demonstrated to reduce the critical micelle concentration (CMC), modulate phase behavior, and promote the formation of tailored micellar structures, thereby optimizing detergent performance. In this study, we employ all-atom molecular dynamics simulations to investigate the morphology and microstructure of single and binary mixed surfactant systems comprising ionic surfactants sodium lauryl ether sulfate (SLES) and linear alkylbenzene sulfonate (LAS) and non-ionic surfactant poly(oxyethylene) n-dodecyl ether (EO7). Our results reveal concentration-dependent transitions in micellar morphology from spherical to ellipsoidal to wormlike-cylindrical aggregates. The addition of non-ionic surfactants reduces electrostatic repulsion between ionic headgroups, leading to larger and more stable aggregates. This results in an increase in the aggregation number by approximately 30% and a corresponding decrease in the solvent-accessible surface area by up to 27%. EO7 exhibits adaptive conformations, compact in ionic mixtures versus extended in single surfactant systems, optimizing solvation and steric interactions. The study highlights critical solvation dynamics in mixed surfactant systems, where the addition of EO7 modulates surface properties by reducing hydrogen bonding between ionic headgroups (SLES/LAS) and water compared to single surfactant systems, while enhancing water–water hydrogen bonding within the first and second solvation shells when the non-ionic concentration is increased. These findings provide molecular-level insights into the self-assembly mechanisms, aggregation behavior, and solvation properties of mixed surfactant systems, contributing to the rational design of optimized formulations for industrial applications.
Read More ...
|
|
|
-
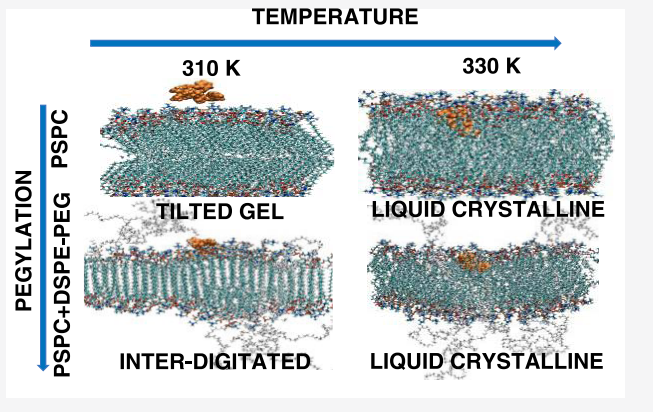
Tamaghna Chakraborti, Riya Sharma, Anand Narayanan Krishnamoorthy, Rakesh Chandran Puthenkalathil, Jayant K Singh “Decoding the Influence of Microstructure on the Stability and Dynamical Properties of the HMT-PMBI Anion Exchange Membrane Using Molecular Dynamics Simulation” The Journal of Physical Chemistry C
Abstract: This work utilizes all-atom simulations to investigate the thermodynamic and transport properties of the hexamethyl-p-terphenyl poly(benzimidazolium) (HMT-PMBI) membrane with chloride anions, focusing on its stability and performance across different temperatures and hydration levels (lambda). By employing generalized AMBER force field (GAFF) and density functional theory (DFT)-derived partial charges, we examine key microstructural features such as the radial distribution function (RDF) and pore size distribution (PSD) and their influence on membrane properties such as density, diffusivity, and conductivity. Our findings show that the cation–anion RDF peak values are reduced by nearly half, and the PSD distribution range expands 2-fold with a 4-fold increase in lambda. This leads to faster decorrelation between cations and anions, as indicated by a significant decrease in cation–anion residence times from 40 ns to less than 0.1 ns, resulting in nearly a 20 times increase in diffusivity and conductivity at higher lambda. Additionally, at low , lambda anions exhibit subdiffusive behavior due to pore sizes smaller than their molecular diameter and the formation of segregated ion conduction channels in multiple clusters rather than a continuous percolated structure. Despite this, the chloride ion conductivity reaches 13 mS/cm at T = 300.15 K and lambda = 20.0, the highest hydration level examined. A comparison with our previous study on Sustainion anion-exchange membrane (AEM) reveals that although HMT-PMBI demonstrates superior chemical stability, its conductivity is lower, mainly due to the cationic group incorporated into the polymer backbone. These insights into the structure–property relationships of anion-exchange membranes (AEMs) are essential for the development of next-generation membranes that balance both durability and performance efficiency.

Read More ...
|
|
|
-
Hariom Birla, Showkat H Mir, Khushboo Yadav, Thomas Halbritter, Alexander Heckel, Jayant K Singh, Thiruvancheril G Gopakumar “Unusual one dimensional cascade effect in the thermal and photo-induced switching of azobenzene derivatives on a graphite surface” Chemical Science
Abstract: Harnessing cooperative switching opens possibilities for engineering the responses of molecular films to external triggers and provides opportunities to control the directionality of switching/reactions and design novel nanostructures. Here, we demonstrate a one dimensional (1D) cascade effect in the thermal- and photo-induced switching of azobenzene derivatives deposited on a graphite surface. Upon thermal- and photo-induction, molecules switch between their geometric states (trans and cis) along a selected lattice within the assembly. We explore the switching at the molecular level using atomic force microscopy (AFM) and scanning tunneling microscopy (STM) and reveal that the 1D cascading effect proceeds along the lattice direction where the inter-molecular interaction is the strongest. Theoretical calculations and experiments reveal a cascading effect of up to 350 molecules for photo-induced and 530 molecules for thermal-induced switching along a given lattice.
Read More ...
|
|
2024
|
|---|
-
Diksha Srivastava, Vipin Mishra, Showkat H Mir, Jyotirban Dey, Jayant K Singh, Manabendra Chandra, Thiruvancheril G Gopakumar “Large Area Film of Highly Crystalline, Cleavable, and Transferable Semi-Conducting 2D-Imine Covalent Organic Framework on Dielectric Glass Substrate” ACS Applied Materials & Interfaces
Abstract: Two dimensional (2D) imine-based covalent organic framework (COF), 2D-COF, is a newly emerging molecular 2D polymer with potential applications in thin film electronics, sensing, and catalysis. It is considered an ideal candidate due to its robust 2D nature and precise tunability of the electronic and functional properties. Herein, we report a scalable facile synthesis of 2D imine-COF with control over film thickness (ranging from 100 nm to a few monolayers) and film dimension reaching up to 2 cm on a dielectric (glass) substrate. Highly crystalline 2D imine polymer films are formed by maintaining a quasi-equilibrium (very slow, 15 h) in Schiff base condensation reaction between p-phenylenediamine (PDA) and benzene-1,3,5-tricarboxaldehyde (TCA) molecules. Free-standing thin and ultrathin films of imine-COF are obtained using sonication exfoliation of 2D-COF polymer. Insights into the microstructure of thin/ultrathin imine-COF are obtained using scanning and transmission electron microscopy (SEM and TEM) and atomic force microscopy (AFM), which shows high crystallinity and 2D layered structure in both thin and ultrathin films. The chemical nature of the 2D polymer was established using X-ray photoelectron spectroscopy (XPS). Optical band gap measurements also reveal a semiconducting gap. This is further established by electronic structure calculation using density functional theory (DFT), which reveals a semiconductor-like band structure with strong dispersion in bands near conduction and valence band edges. The structural characteristics (layered morphology and microscopic structure) of 2D imine-COF show significant potential for its application in thin film device fabrication. In addition, the electronic structure shows strong dispersion in the frontier bands, making it a potential semiconducting material for charge carrier transportation in electronic devices.

Read More ...
|
|
|
|
|
|
|
-
Bokinala Moses Abraham, Mullapudi V Jyothirmai, Priyanka Sinha, Francesc Viñes, Jayant K Singh, Francesc Illas “Catalysis in the digital age: Unlocking the power of data with machine learning” Wiley Interdisciplinary Reviews: Computational Molecular Science
Abstract: The design and discovery of new and improved catalysts are driving forces for accelerating scientific and technological innovations in the fields of energy conversion, environmental remediation, and chemical industry. Recently, the use of machine learning (ML) in combination with experimental and/or theoretical data has emerged as a powerful tool for identifying optimal catalysts for various applications. This review focuses on how ML algorithms can be used in computational catalysis and materials science to gain a deeper understanding of the relationships between materials properties and their stability, activity, and selectivity. The development of scientific data repositories, data mining techniques, and ML tools that can navigate structural optimization problems are highlighted, leading to the discovery of highly efficient catalysts for a sustainable future. Several data-driven ML models commonly used in catalysis research and their diverse applications in reaction prediction are discussed. The key challenges and limitations of using ML in catalysis research are presented, which arise from the catalyst's intrinsic complex nature. Finally, we conclude by summarizing the potential future directions in the area of ML-guided catalyst development.

Read More ...
|
-
Priyanka Sinha, MV Jyothirmai, B Moses Abraham, Jayant K Singh “Machine learning driven advancements in catalysis for predicting hydrogen evolution reaction activity” Materials Chemistry and Physics
Abstract: In the field of catalysis research, the emergence of machine learning (ML) has triggered a significant transformation, revolutionizing our methodologies for exploring and comprehending the dynamics of hydrogen evolution reaction (HER) activity. This review explores the burgeoning adoption of ML techniques in catalysis research, with a particular focus on their application in predicting HER activity. The review begins with an introduction to the ML workflow and its relevance in predicting catalytic performance. Emphasis is given to the significance of data quality and quantity, highlighting the need for well-defined input variables and the continuous evolution of catalysis-specific databases. We also accentuates the pivotal role of descriptors in utilizing ML for HER activity prediction, emphasizing the importance of proper selection based on database size and features to capture domain knowledge of diverse material properties. Furthermore, this review comprehensively examines the application of ML techniques in the context of HER, enabling accurate predictions of catalytic performance. Finally, the review explores immediate research needs and outlines future directions in this rapidly evolving field.
Read More ...
|
|
|
-
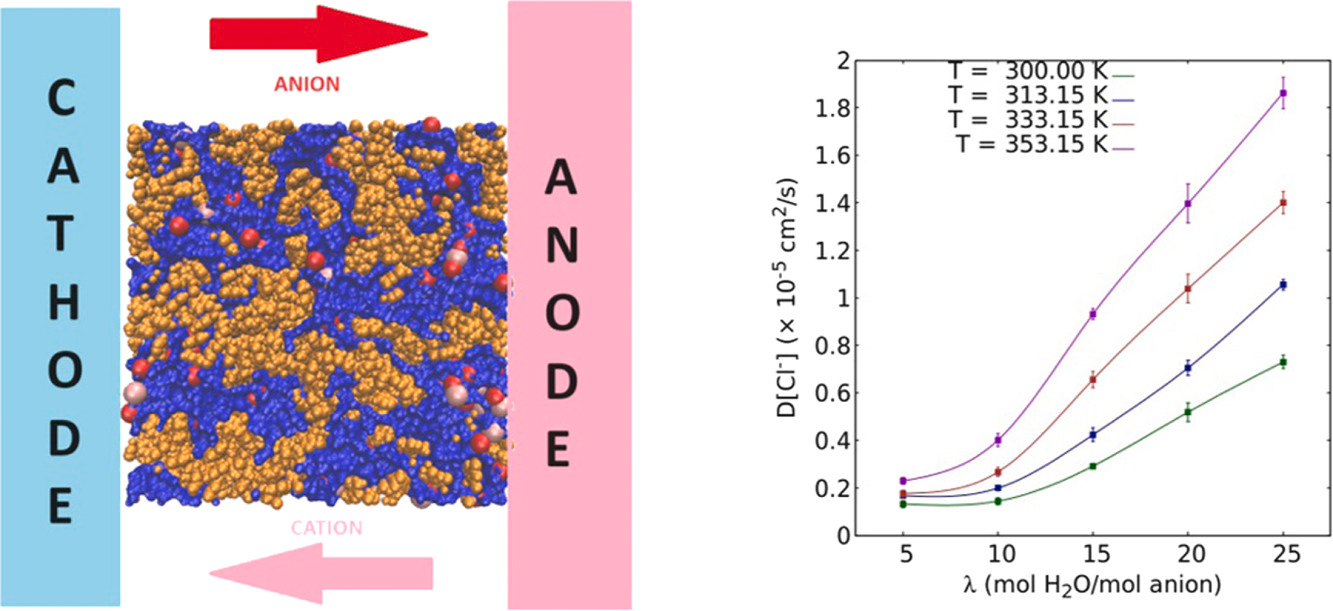
Tamaghna Chakraborti, Riya Sharma, Anand Narayanan Krishnamoorthy, Harshal Chaudhari, Kuldeep Mamtani, Jayant K Singh “Unravelling the effect of molecular interactions on macroscale properties in Sustainion anion exchange membrane (AEM) under hydrated conditions using MD simulations” Journal of Membrane Science
Abstract: Sustainion is an anion exchange water electrolyzer membrane that has exhibited a scalable performance. In this work, we present a molecular model for the Sustainion membrane incorporating functionalization of the polymer in order to better mimic experimental conditions. Here in, we present a comprehensive exploration of its structural and transport properties like density, diffusivity and conductivity at various operating hydration levels using molecular dynamics simulations. The density exhibits a non-monotonic trend while the diffusivity showcases a non-linear behaviour with hydration. Furthermore, diffusion exhibits an Arrhenius-like dependence on temperature with the activation energies exhibiting a non-monotonic relationship akin to the density. It is concluded that the afore-mentioned properties of the Sustainion membrane are due to the counteracting influence of the enhancement in coordination number, and a reduction in the potentials of mean force of the different atomic pairs in the system. The effect of added salt on Sustainion properties is also determined and collated with experiments. Based on comparison with experiment, we deduce that vehicular diffusion is the predominant mechanism in the diffusion of the chloride ion, while it contributes approximately 15pc to the diffusivity of the hydroxide ion.
Read More ...
|
-
Diksha Srivastava, Vipin Mishra, Showkat H Mir, Jyotirban Dey, Jayant K Singh, Manabendra Chandra, Thiruvancheril G Gopakumar “Large Area Film of Highly Crystalline, Cleavable, and Transferable Semi-Conducting 2D-Imine Covalent Organic Framework on Dielectric Glass Substrate” ACS Applied Materials & Interfaces
Abstract: Two dimensional (2D) imine-based covalent organic framework (COF), 2D-COF, is a newly emerging molecular 2D polymer with potential applications in thin film electronics, sensing, and catalysis. It is considered an ideal candidate due to its robust 2D nature and precise tunability of the electronic and functional properties. Herein, we report a scalable facile synthesis of 2D imine-COF with control over film thickness (ranging from 100 nm to a few monolayers) and film dimension reaching up to 2 cm on a dielectric (glass) substrate. Highly crystalline 2D imine polymer films are formed by maintaining a quasi-equilibrium (very slow, 15 h) in Schiff base condensation reaction between p-phenylenediamine (PDA) and benzene-1,3,5-tricarboxaldehyde (TCA) molecules. Free-standing thin and ultrathin films of imine-COF are obtained using sonication exfoliation of 2D-COF polymer. Insights into the microstructure of thin/ultrathin imine-COF are obtained using scanning and transmission electron microscopy (SEM and TEM) and atomic force microscopy (AFM), which shows high crystallinity and 2D layered structure in both thin and ultrathin films. The chemical nature of the 2D polymer was established using X-ray photoelectron spectroscopy (XPS). Optical band gap measurements also reveal a semiconducting gap. This is further established by electronic structure calculation using density functional theory (DFT), which reveals a semiconductor-like band structure with strong dispersion in bands near conduction and valence band edges. The structural characteristics (layered morphology and microscopic structure) of 2D imine-COF show significant potential for its application in thin film device fabrication. In addition, the electronic structure shows strong dispersion in the frontier bands, making it a potential semiconducting material for charge carrier transportation in electronic devices.
Read More ...
|
|
|
-
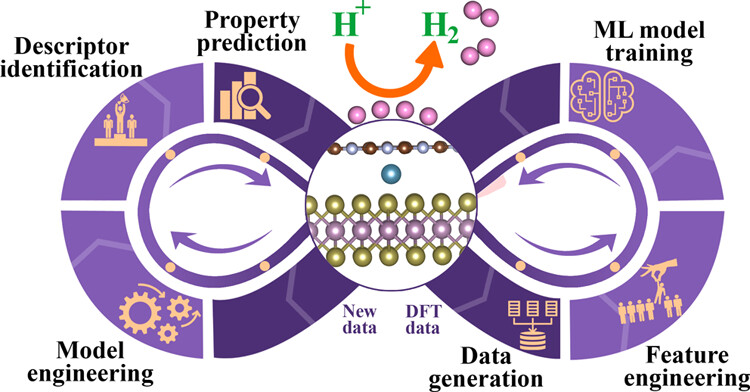
MV Jyothirmai, Roshini Dantuluri, Priyanka Sinha, B. Moses Abraham “Machine Learning Driven High-Throughput Screening of Transition Metal Atom Intercalated g-C3N4/MX2 (M = Mo, W; X = S, Se, Te) Heterostructures for Hydrogen Evolution Reaction” ACS Applied Materials and Interfaces
Abstract: Rising global energy demand, accompanied by environmental concerns linked to conventional fossil fuels, necessitates a shift toward cleaner and sustainable alternatives. This study focuses on the machine-learning (ML)-driven high-throughput screening of transition-metal (TM) atom intercalated g-C3N4/MX2 (M = Mo, W; X = S, Se, Te) heterostructures to unravel the rich landscape of possibilities for enhancing the hydrogen evolution reaction (HER) activity. The stability of the heterostructures and the intercalation within the substrates are verified through adhesion and binding energies, showcasing the significant impact of chalcogenide selection on the interaction properties. Based on hydrogen adsorption Gibbs free energy (del GH) computed via density functional theory (DFT) calculations, several ML models were evaluated, particularly random forest regression (RFR) emerges as a robust tool in predicting HER activity with a low mean absolute error (MAE) of 0.118 eV, thereby paving the way for accelerated catalyst screening. The Shapley Additive exPlanation (SHAP) analysis elucidates pivotal descriptors that influence the HER activity, including hydrogen adsorption on the C site (HC), MX layer (HMX), S site (HS), and intercalation of TM atoms at the N site (IN). Overall, our integrated approach utilizing DFT and ML effectively identifies hydrogen adsorption on the N site (site-3) of g-C3N4 as a pivotal active site, showcasing exceptional HER activity in heterostructures intercalated with Sc and Ti, underscoring their potential for advancing catalytic performance.
Read More ...
|
-
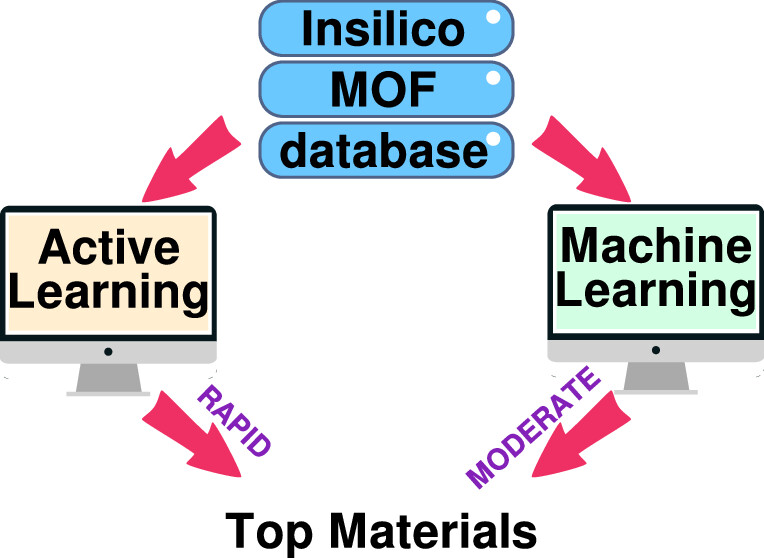
Varad Dao, Jayant K. Singh “Accelerating insilico discovery of metal-organic frameworks for ethane/ethylene and propane/propylene separation: a synergistic approach integrating molecular simulation, machine learning, and active learning” ACS Applied Materials and Interfaces
Abstract: Cryogenic distillation, a currently employed method for C2H4/C2H6 and C3H6/C3H8 mixture separation, is energy-intensive, prompting the research toward alternative technologies, including adsorbent-based separation. In this work, we combine machine learning (ML) technique with high-throughput screening to screen 23,000 hypothetical metal–organic frameworks (MOFs) for paraffin (C2H6 and C3H8) selective adsorbent separation. First, structure-based prescreening was employed to remove MOFs with undesired geometric properties. Further, a random forest model built upon the multicomponent grand canonical Monte Carlo (m-GCMC) simulation data of training set MOFs was found to be the most successful in learning the relationship between MOF features and olefin/paraffin mixture separation. Using this technique, the separation performance of the remaining (test set) MOFs was predicted, and the top-performing MOFs were identified. We also employed active learning (AL) to evaluate its effectiveness in improving the prediction of olefin/paraffin selectivity. AL was discovered to be 29 times more efficient than the best-supervised ML model, as it was able to identify the top materials in limited training data and at a fraction of computational cost and time as compared to ML techniques. Among the top selected materials, framework chemistry was found to be the most important parameter. Nickel and copper (as a metal node) in a tfzd and hms topological arrangement respectively, were discovered to be a prevalent attribute in high-performing MOFs, further demonstrating the prominent significance of framework chemistry. Additionally, the top MOFs discovered were studied in detail and further compared to the previously reported MOFs. These MOFs show the highest selectivity for C2H4/C2H6 and C3H6/C3H8 mixture separation, as reported until date. The hierarchical strategy devised in this study will facilitate the quick screening of MOFs across multiple databases toward industrially significant separation processes by leveraging molecular simulations and AL.
Read More ...
|
|
2023
|
|---|
-
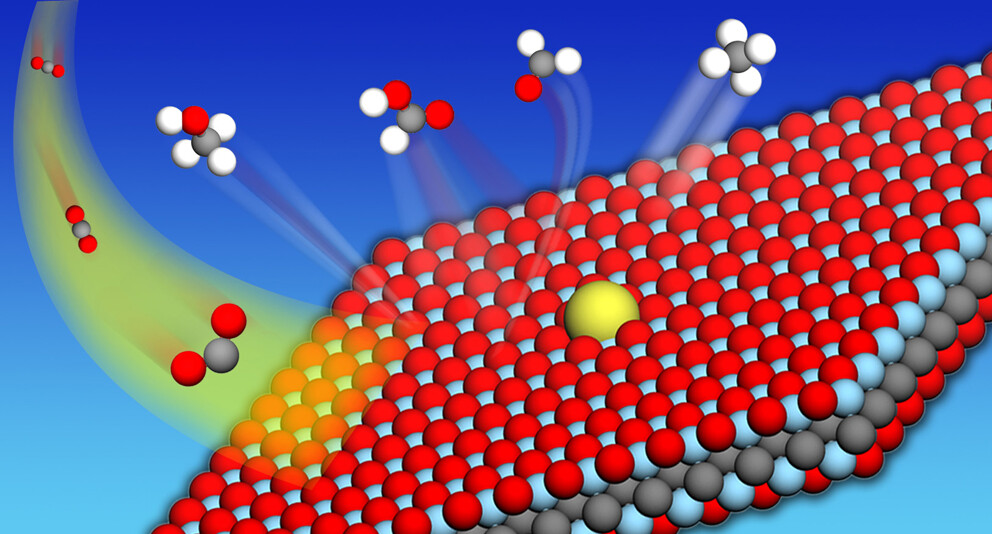
Ajinkya Athawale, B Moses Abraham, MV Jyothirmai, Jayant K Singh “MXene-Based Single-Atom Catalysts for Electrochemical Reduction of CO2 to Hydrocarbon Fuels” The Journal of Physical Chemistry C
Abstract: The sustainable and cost-effective reduction of electrochemical CO2 to valuable chemicals or fuels is a promising solution to mitigate greenhouse gas emissions and energy demands. Herein, the potential of MXenes as an anchoring site for isolated transition metal (TM) atoms is explored to develop efficient single-atom catalysts (SACs) for the electrochemical CO2 reduction reaction (CO2RR). We design a series of SACs from 3d (Sc, Ti, V, Cr, Mn), 4d (Y, Zr, Nb, Mo), and 5d (Hf) transition metals, supported on an O-terminated MXene (TM@Ti2CO2) using well-defined first-principles calculations. Our results show that the TMs anchored on top of the carbon atom of Ti2CO2 (hollow-C site) exhibit the most stable configuration. The electronic calculations demonstrate a strong correlation between adsorption energy and various chemical properties such as average bond distances (dTM–O), Bader charge, work function, and d-electron center of the metal, suggesting that the complex interplay between the electronic and geometric properties of the adsorbing atom can serve as descriptors for determining the adsorption energy. The filling of d-orbitals influences the degree of charge transfer by creating an attractive interaction between the CO2RR intermediate species and single TM atoms with a positive charge, promoting efficient catalytic CO2 reduction through charge-induced dipole interactions. Particularly, the Ti atom anchored on Ti2CO2 exhibited the most favorable performance as a catalyst for the CO2RR, exhibiting the lowest limiting potential among the SACs examined. Moreover, most of the examined SACs showed selectivity toward the CO2RR over the hydrogen evolution reaction by comparing the changes in the Gibbs free energy of the first hydrogenation step. Our study offers valuable insights for developing MXene-based SACs for the CO2RR, paving the way for efficient electrocatalyst design in the future.
Read More ...
|
-
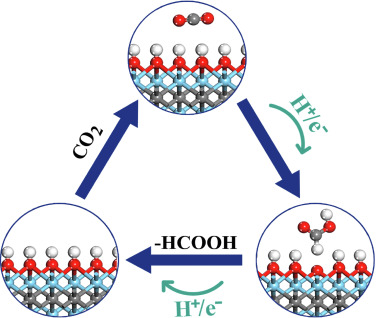
Vanshree Parey, B Moses Abraham, Jayant K Singh “Surface hydroxylation mechanism of Zr2X(OH)2 (X = C, N, or B) MXenes: A promising catalyst for CO2 conversion into formic acid” Materials Chemistry and Physics
Abstract: Electrocatalytic transformation of CO2 to formic acid is typically realized via bi-hydrogenation process, which is remarkably challenging because of high thermodynamic stability and chemical inertness of the intermediate. Here, we explored the surface hydroxylation mechanism for CO2 conversion into formic acid over OH-terminated MXenes that can effectively circumvent the thermodynamic sink of the conventional bi-hydrogenation process by capturing additional hydrogen from the surface. The observed unique CO2 RR activity can be attributed to the reactive H atom on the OH-terminated MXenes. The facile capture of (H) from OH-terminated MXenes confirm the viability of surface-hydroxylation mechanism. In addition, the ultra low work functions (1.72–2.27 eV) of such materials demonstrate the significance of functional groups in deciding the surface electrostatic potential of MXenes during the etching process. Overall, the present study bypass the thermodynamic constraints hampering the formation of formic acid by eliminating the intermediate step in the CO2 hydrogenation reaction and is likely to have considerable implications in the fields of catalytic chemistry and CO2 conversion.
Read More ...
|
-
B Moses Abraham, MV Jyothirmai, Jayant K Singh “Electrochemical CO2 Conversion via MXenes: A DFT Perspective” Age of MXenes, Volume 1. Fundamentals and Artificial Intelligence: Machine Learning Interventions
Abstract: The electrochemical conversion of carbon dioxide (CO2) to produce clean fuels and high-value chemicals have received great attention for the generation of green and sustainable energy. Developing highly efficient and low cost electrocatalysts mainly depends on fundamental understanding of catalytic mechanisms and their structure activity relationships. Recently, density functional theory (DFT) has significantly transformed our fundamental understanding of atomic scale details to develop these relationships for revealing the possible mechanisms involved in the catalytic reactions. In this chapter, we briefly summarize the application of DFT in electrocatalysis to analyze the reaction mechanisms in transition metal carbides/nitrides (also known as MXenes) for CO2 conversion into
green fuels. Some useful tools for theoretical analysis were highlighted for evaluating the electrocatalytic performances. The potential ability of MXenes to capture, activate, and dissociate CO2 is mainly due to Dewar interactions involving hybridization between d orbitals of metals and CO2 pi orbitals. The catalytic selectivity of MXenes towards CO2 conversion is higher than hydrogen evolution reaction (HER), signifying the efficiency of the catalyst for CO2RR. Overall, the theoretical findings discussed in the present chapter can provide useful insights to rationalize the development of MXene based electrocatalysts for CO2RR into renewable fuels and chemicals.

Read More ...
|
|
|
-
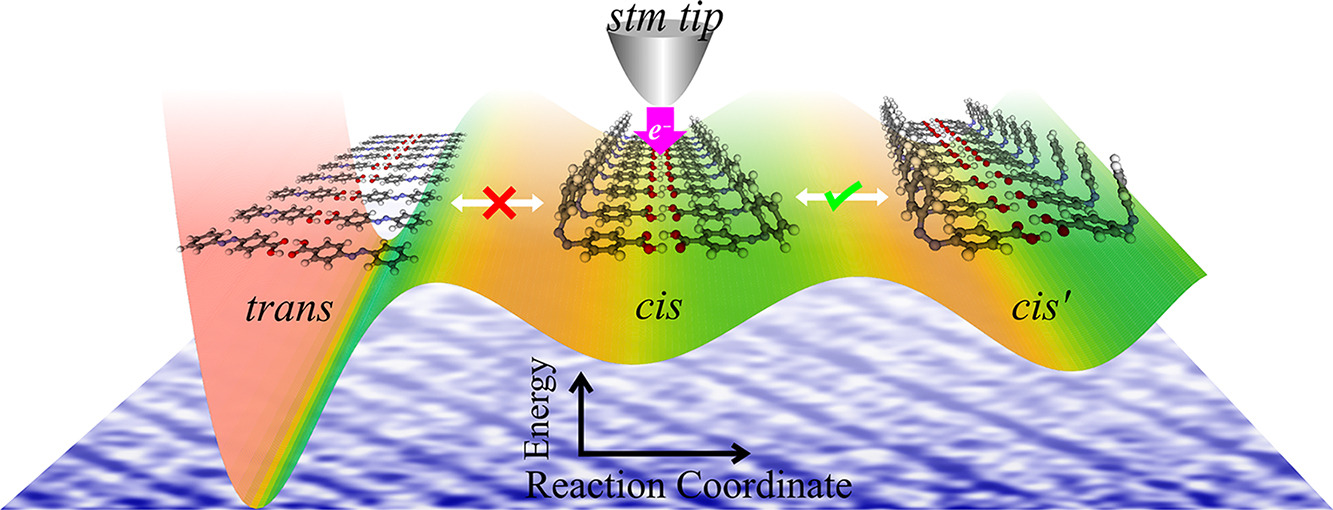
Hariom Birla, Showkat H Mir, Khushboo Yadav, Thomas Halbritter, Alexander Heckel, Jayant K Singh, Thiruvancheril G Gopakumar “Tuning the Switching Probability of Azobenzene Derivatives on Graphite Surface through Chemical Functions” The Journal of Physical Chemistry C
Abstract: Molecules that can be triggered between different states, molecular switches, are considered as the building blocks for molecular electronics. The chemical nature of the molecular switches is decisive in controlling the mechanism/energy barrier of switching and the life time of different states. Here, we investigate the electronic structure, switching barrier, and electron/hole-induced switching of an adlayer of three different azobenzene (AB) derivatives on graphite surface. The adlayers of AB derivatives with carboxyl group form a hydrogen-bonded dimer-based assembly and the derivative with the thiocyanate group forms van der Waals-stabilized assembly. While all the molecules in the adlayer can be individually switched using electron/hole, the switching probability depends on their chemical nature. We use a combination of scanning tunneling microscopy and concomitant density functional theory calculations to demonstrate the switching probability of different AB derivatives. Molecules that are having strong inter-molecular interactions within the adlayer show low switching probability compared to the one having weak inter-molecular interaction. Additionally, we observe that the molecule–surface interaction also contributes to the switching.
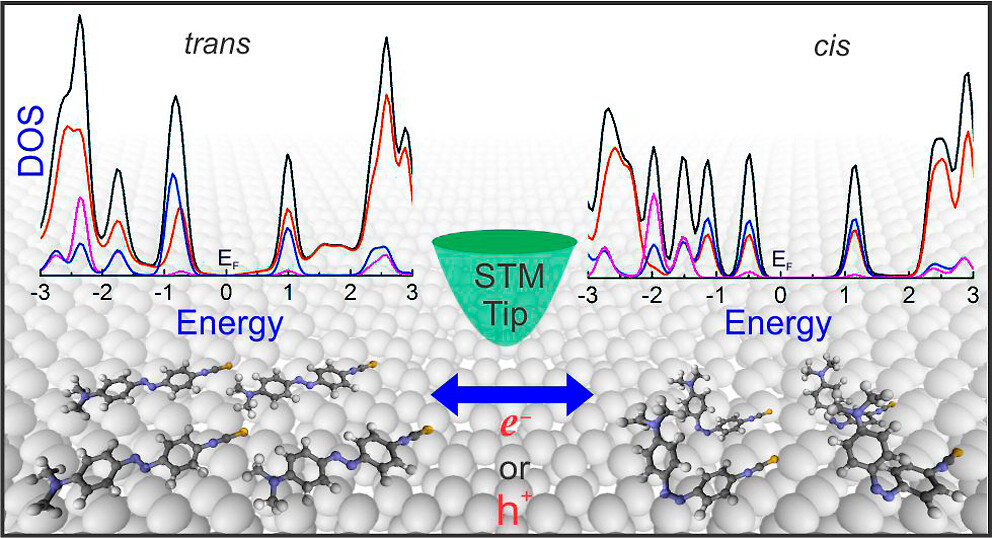
Read More ...
|
-
Priyanka Sinha, D Roshini, Varad Daoo, B Moses Abraham, Jayant K Singh “Integrating Machine Learning and Molecular Simulation for Material Design and Discovery” Transactions of the Indian National Academy of Engineering
Abstract: Machine learning (ML) and artificial intelligence (AI) have enabled transformative impact on materials science by accelerating cutting-edge insights from computational methods and their analysis to hitherto unattainable scales. Such an assembly of linear algebra and statistical methods can facilitate the conceptual development of flexible techniques by finding mechanism/information/hidden pattern in a data set. The present review provides basic information about the classification of ML methodology and its workflow. These sections also elaborate on the advantages and limitations of various ML algorithms for solving problems in materials science and reviewing cases of success and failure. Subsequently, we show how these techniques can uncover the complexities in several quantitative structure–property relationships to design and discover novel materials for various applications. We conclude our review with an outlook on present research challenges, problems, and potential future perspectives in the field of machine learning. Overall, this review can serve as a fundamental guide to amplify the adoption of such tools and methods by materials scientists across academia and industry.
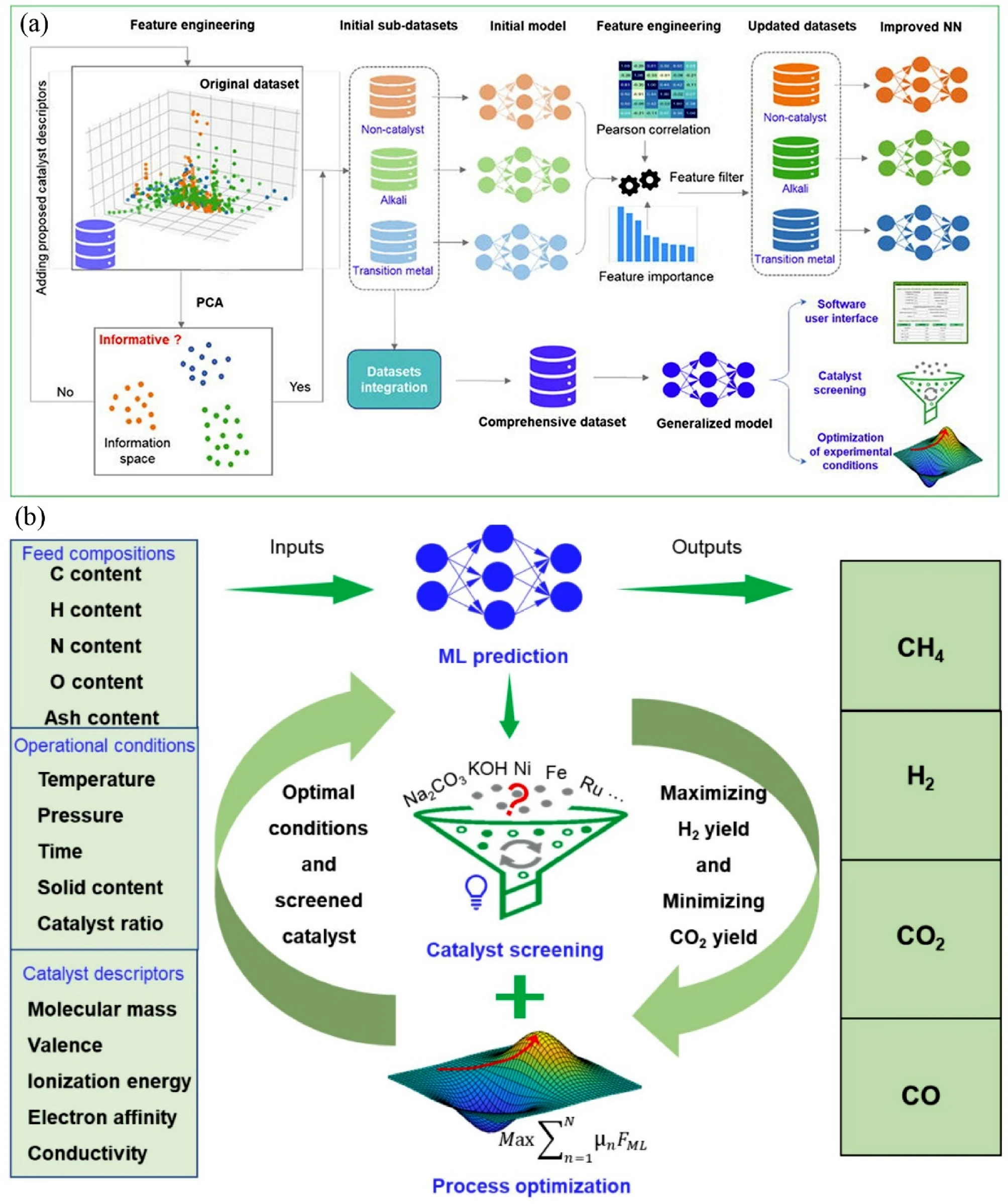
Read More ...
|
|
|
-
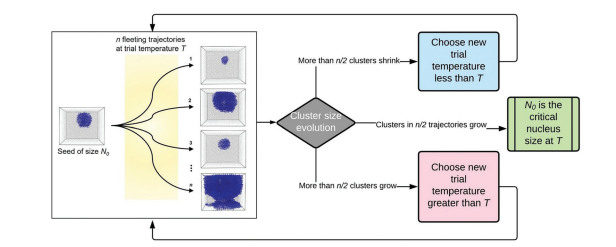
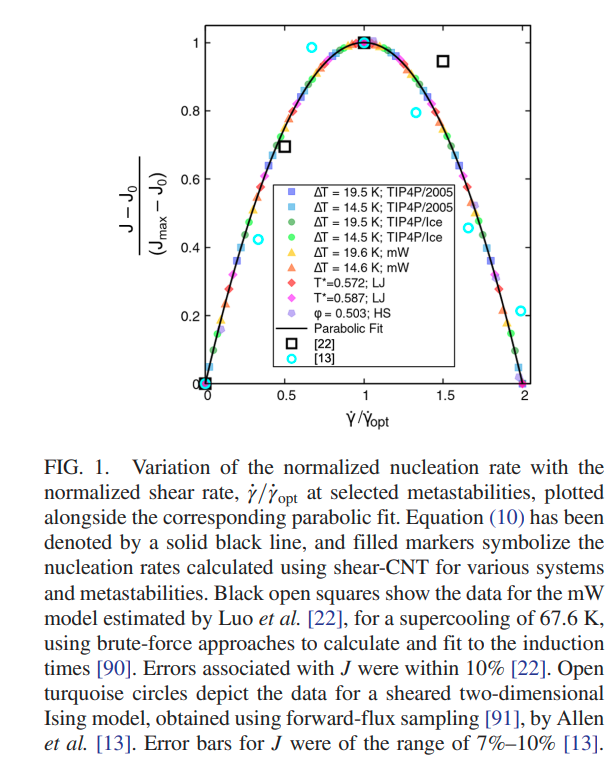
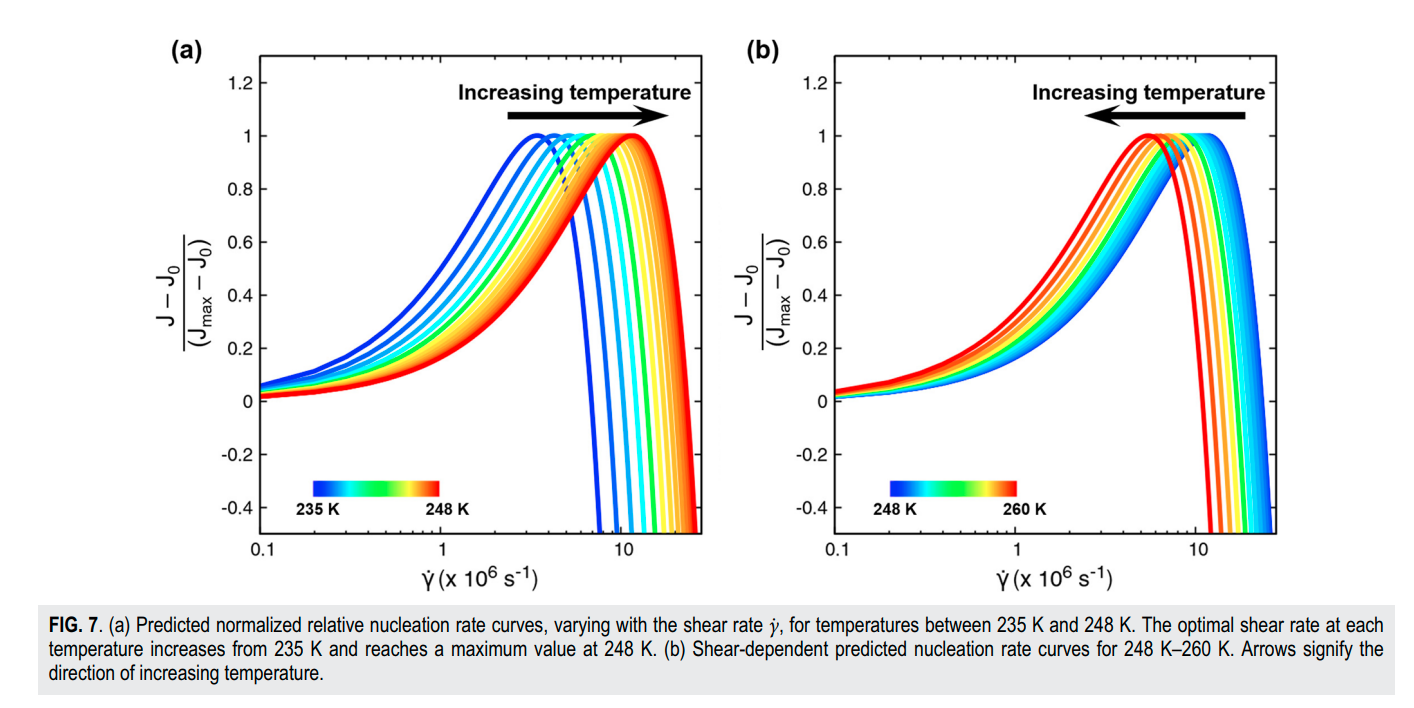
Srirangam, Snehitha; Bhendale, Mangesh; Singh, Jayant “Does supercooled water retain its universal nucleation behavior under shear at high pressure?” Physical Chemistry Chemical Physics
Abstract: Understanding the nucleation of homogeneous flow systems at high pressures is vital in protein crystallization and cryopreservation, where high pressure prevents the freezing of biological samples. This study examines the behavior of ice nucleation under shear at various pressures and explores the universal nucleation behavior of the sheared systems applied to supercooled water at higher pressures. In this study, the nucleation rates for TIP4P/Ice model via the seeding method based on extended classical nucleation theory (CNT) are computed at pressures of 1, 100, 500, 700, and 1000 bar and a constant temperature of 240 K. Using extended CNT with explicitly embodying the shear rate, we analyzed the dependence of pressure on the transport and thermodynamic properties. In line with the previous studies, we observed that the chemical potential difference between ice and liquid and viscosity decrease while diffusivity increases with an increase in pressure. Furthermore, we showed that the dependence of nucleation rate with shear at higher pressure is non-monotonic, with the maximum at optimal shear rates between 107 to 108 /s . Our results demonstrate a non-monotonic pressure dependence of the optimal shear rates, which could originate from a violation of the Stokes-Einstein relation.
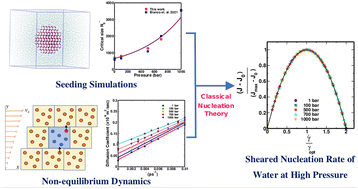
Read More ...
|
-
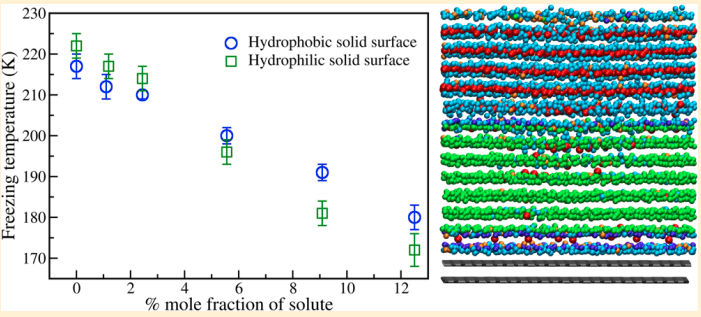
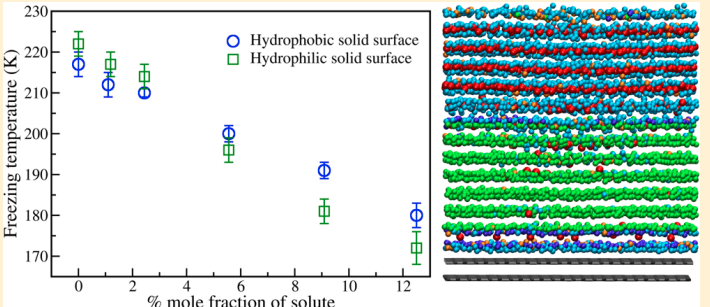
Indra, Aindrila; Bhendale, Mangesh; Singh, Jayant “Understanding the role of polymers on the nucleating behavior of water in dilute supercooled solutions” The Journal of Chemical Physics
Abstract: Understanding the nucleation behavior of water in dilute polymeric solutions is quintessential for the development of suitable artificial ice recrystallization inhibition (IRI) agents. Although poly(vinyl alcohol) (PVA) is found to be one of the most potent biomimetic IRI agents, the molecular understanding of the nucleation behavior of water in the presence of PVA is still lacking. Here, we use molecular dynamics to elucidate the role of concentration, degree of supercooling, degree of polymerization, and amphiphilicity of PVA and PVA-like polymers on the homogeneous nucleation of water in dilute polymeric solutions using the seeding method. Using classical nucleation theory (CNT), our simulations indicate an increase in the chemical potential difference between ice and melt that favors ice nucleation. However, it also predicts a significant increase in the ice–melt interfacial energy that impedes nucleation. The relative increase in the interfacial energy dominates the increase in the chemical potential difference, which results in a decrease in the nucleation rate of water with an increase in the solute concentration. This study contradicts the previous simulation study that suggested the promotion of homogeneous ice nucleation by PVA and supports the experimental observations of the heterogeneous origins of ice nucleation. Our results also suggest the non-classical origins of ice nucleation in polymeric solutions and the limitation of the CNT in predicting heterogeneous ice nucleation in polymeric solutions.
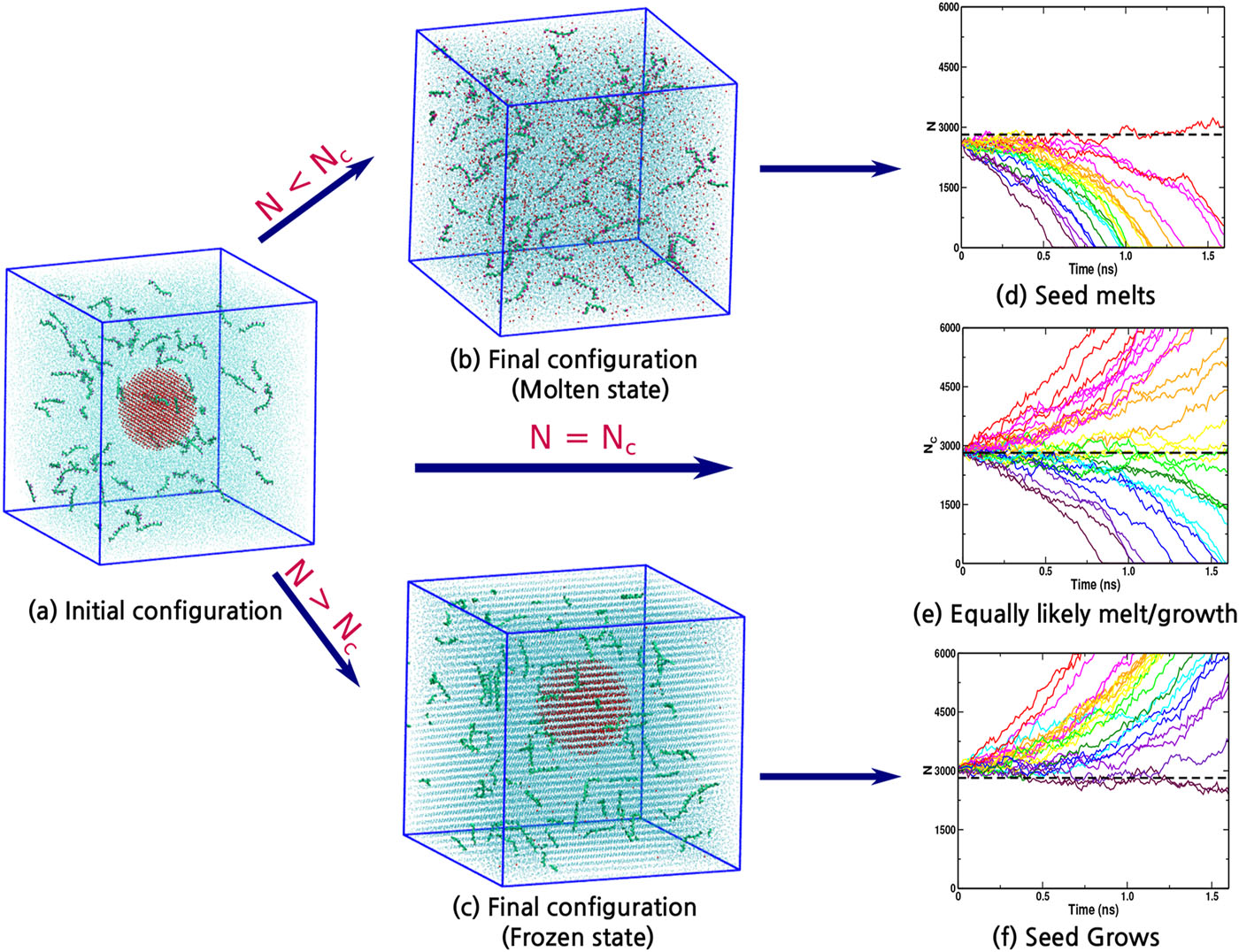
Read More ...
|
-
B Moses Abraham, Oriol Piqué, Mohd Aamir Khan, Francesc Viñes, Francesc Illas, Jayant K Singh “Machine Learning-Driven Discovery of Key Descriptors for CO2 Activation over Two-Dimensional Transition Metal Carbides and Nitrides” ACS Applied Materials & Interfaces
Abstract: Fusing high throughput quantum mechanical screening techniques with modern artificial intelligence strategies is among the most fundamental yet revolutionary science activities, capable of opening new horizons in catalyst discovery. Here, we apply this strategy to the process of finding appropriate key descriptors for CO2 activation over two dimensional transition metal (TM) carbides/nitrides (MXenes). Various machine learning (ML) models are developed to screen over 114 pure and defective MXenes, where the random forest regressor (RFR) ML scheme exhibits the best predictive performance for the CO2 adsorption energy, with a mean absolute error +/- standard deviation of 0.16 +/- 0.01 and 0.42 +/- 0.06 eV for training and test data sets, respectively. Feature importance analysis revealed d band center (eps d), surface metal electronegativity (chi M), and valence electron number of metal atoms (MV) as key descriptors for CO2 activation. These findings furnish a fundamental basis for designing novel MXene based catalysts through the prediction of potential indicators for CO2 activation and their posterior usage.
Read More ...
|
|
|
|
|
|
|
-
Abraham, B. Moses and Sinha, Priyanka and Halder, Prosun and Singh, Jayant K. “Fusing a machine learning strategy with density functional theory to hasten the discovery of 2D MXene-based catalysts for hydrogen generation” Journal of Materials Chemistry A
Abstract: The complexity of the topological and combinatorial configuration space of MXenes can give rise to gigantic design challenges that cannot be addressed through traditional experimental or routine theoretical methods. To this end we establish a robust and more broadly applicable multistep workflow using supervised machine learning (ML) algorithms to construct well trained data driven models for predicting the hydrogen evolution reaction (HER) activity of 4500 MMXT2 type MXenes where 25 of the materials space (1125 systems) is randomly selected to evaluate the HER performance using density functional theory (DFT) calculations. As the most desirable ML model the gradient boosting regressor (GBR) processed with recursive feature elimination (RFE) hyperparameter optimization (HO) and the leave one out (LOO) approach accurately and rapidly predicts the Gibbs free energy of hydrogen adsorption (delG) with a low predictive mean absolute error (MAE) of 0.358 eV. Based on these observations the H atoms adsorbed directly on top of the outermost metal atom layer of the MMXT2 type MXenes (site 1) with Nb Mo and Cr metals with O functionalization are discovered to be highly stable and active for catalysis surpassing commercially available platinum based counterparts. Overall the physically meaningful predictions and insights of the developed ML and DFT based multistep workflow will open new avenues for accelerated screening rational design and discovery of potential HER catalysts.
Read More ...
|
|
|
-
Bhendale, Mangesh and Singh, Jayant K “Molecular Insights on Morphology, Composition, and Stability of Mixed Micelles Formed by Ionic Surfactant and Nonionic Block Copolymer in Water Using Coarse-Grained Molecular Dynamics Simulations” Langmuir
Abstract: The nanoscale association domains are the ultimate determinants of the macroscopic properties of complex fluids involving amphiphilic polymers and surfactants, and hence, it is foremost important to understand the role of polymer/surfactant concentration on these domains. We have used coarse-grained molecular dynamics simulations to investigate the effect of polymer/surfactant concentration on the morphology of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) (PEO-PPO-PEO, i.e., pluronics or poloxamers) block copolymer, and ionic surfactants sodium dodecyl sulfate (SDS), mixed micelles in aqueous solution. The proclivity of the surfactant to form the mixed micelles is also probed using umbrella sampling simulations. In this study, we observed that the core of the pluronic + SDS formed mixed micelles consists of PPO, the alkyl tail of SDS, and some water molecules, whereas the PEO, water, and sulfate headgroups of SDS form a shell, consistent with experimental observations. The micelles are spherical at high-pluronic/low-SDS compositions, ellipsoidal at high-SDS/low-pluronic compositions, and wormlike-cylindrical at high-pluronic/high-SDS compositions. The transitions in micelle morphology are governed by the solvent accessible surface area of mixed aggregates, electrostatic repulsion between SDS-headgroups, and dehydration of PEO and PPO segments. The free energy barrier for SDS escape is much higher in mixed micelles than in pure SDS micelles, indicating a stronger tendency for SDS to form pluronic-SDS mixed micelles.
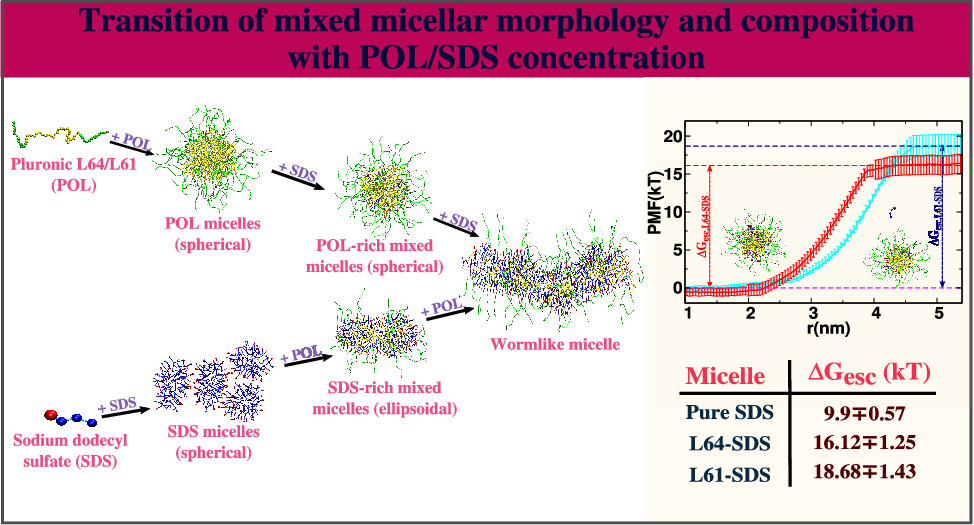
Read More ...
|
|
2022
|
|---|
-
Jyothirmai, M. V. and Abraham, B. Moses and Singh, Jayant K. “The pressure induced phase diagram of double-layer ice under confinement: a first-principles study” Phys. Chem. Chem. Phys.
Abstract: Here, we present double-layer ice confined within various carbon nanotubes (CNTs) using state-of-the-art pressure induced (5 GPa to 5 GPa) dispersion corrected density functional theory (DFT) calculations. We find that the double-layer ice exhibits remarkably rich and diverse phase behaviors as a function of pressure with varying CNT diameters. The lattice cohesive energies for various pure double layer ice phases follow the order of hexagonal pentagonal square tube hexagonal close packed (HCP) square buckled rhombic (b RH). The confinement width was found to play a crucial role in the square and square tube phases in the intermediate pressure range of about 0 to1 GPa. Unlike the phase transition in pure bilayer ice structures, the relative enthalpies demonstrate that the pentagonal phase, rather than the hexagonal structure, is the most stable ice polymorph at ambient pressure as well as in a deep negative pressure region, whereas the b RH phase dominates under high pressure. The relatively short O distance of b RH demonstrates the presence of a strong hydrogen bonding network, which is responsible for stabilizing the system.
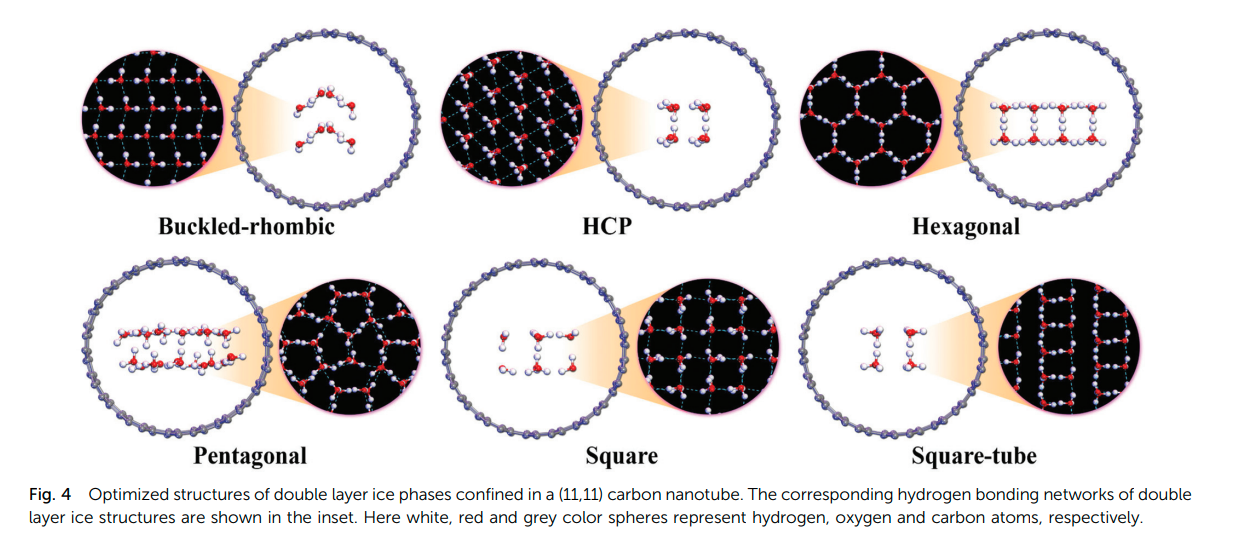
Read More ...
|
-
Mir, Showkat; Yadav, Vivek; Singh, Jayant “Efficient CO2 capture and activation on novel two-dimensional transition metal borides” ACS Applied Materials & Interfaces
Abstract: The large-scale production of CO2 in the atmosphere has triggered global warming, the greenhouse effect, and ocean acidification. The CO2 conversion to valuable chemical products or its capture and storage are of fundamental importance to mitigate the greenhouse effect on the environment. Therefore, exploring new two-dimensional (2D) materials is indispensable due to their potential intriguing properties. Here, we report a new family of 2D transition metal borides (M2B2, M = Sc, Ti, V, Cr, Mn, and Fe; known as MBenes) and demonstrate their static and dynamic stability. These MBenes show a metallic nature and exhibit excellent electrical conductivity. The CO2 adsorption energy on MBenes ranges from 1.04 to 3.95 eV and exhibits the decreasing order as Sc2B2, Ti2B2, V2B2, Cr2B2, Mn2B2, Fe2B2. The spinpolarization calculation shows a reduction in the adsorption energy for magnetic systems. Bader charge transfer indicates the formation of CO2 moiety on the MBene surface, so-called activated CO2, which is essential for its reaction with other surface chemicals. Differential charge density plots reveal a significant charge accumulation around the CO2 molecule. Our theoretical results predict the usage of new MBenes as a cost-effective catalyst for CO2 capture and activation
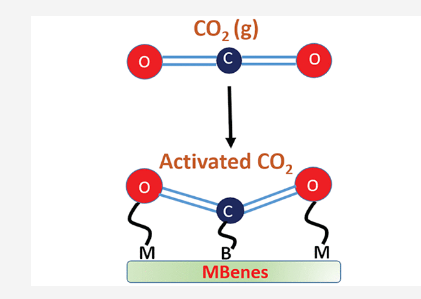
Read More ...
|
-
B. Moses Abraham, Vanshree Parey, Jayant K. Singh “A strategic review of MXenes as emergent building blocks for future two-dimensional materials: recent progress and perspectives” Journal of Materials Chemistry C
Abstract: The breathtaking success of MXenes arising from a library of unique and fascinating properties has triggered world-wide research interest and opened up several new directions in understanding the science and technology of two-dimensional materials. This review provides a holistic evaluation of relevant properties across a wide range of MXene families. The key points of modern approaches applied to MXenes are presented with special emphasis to cover their pitfalls, peculiarities and applications. By considering experimental and theoretical studies, we describe the fundamental structure–property–performance inter-relationships with the goal of providing a better understanding of these MXenes at the atomic level. A final note on immediate research needs along with future directions and major challenges in developing these MXenes is discussed.
Read More ...
|
|
|
-
Bhendale, Mangesh; Srivastava, Arpita; Singh, Jayant “Insights into the Phase Diagram of Pluronic L64 Using Coarse-Grained Molecular Dynamics Simulations” The Journal of Physical Chemistry
Abstract: We investigate the concentration-dependent phase diagram of pluronic L64 in aqueous media at 300 and 320 K using coarse-grained (CG) molecular dynamics (MD) simulations. The CG model is derived by adapting the Martini model for nonbonded interactions along with the Boltzmann inversion (BI) of bonded interactions from all-atom (AA) simulations. Our derived CG model successfully captures the experimentally observed micellar-, hexagonal-, lamellar-, and polymer-rich solution phase. The end-to-end distance reveals the conformational change from an open-chain structure in the micellar phase to a folded-chain structure in the lamellar phase, increasing the orientational order. An increase in temperature leads to expulsion of water molecules from the L64 moiety, suggesting an increase in L64 hydrophobicity. Thermodynamic analysis using the two-phase thermodynamics (2PT) method suggests the entropy of the system decreases with increasing L64 concentration and the decrease in free energy (F) with temperature is mainly driven by the entropic factor (TS). Further, the increase in aggregation number at lower concentrations and self-assembly at very high concentrations is energetically driven, whereas the change from the micellar phase to the lamellar phase with increasing L64 concentration is entropically driven. Our model provides molecular insights into L64 phases which can be further explored to design functionality-based suprastructures for drug delivery and tissue engineering applications.
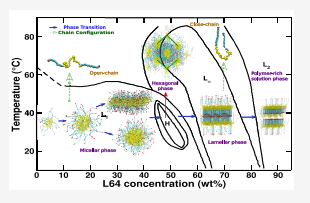
Read More ...
|
-
Debabrata Pramanik, Aiswarya B. Pawar, Sudip Roy, Jayant Kumar Singh “Mechanistic insights of key host proteins and potential repurposed inhibitors regulating SARS-CoV-2 pathway” Journal of Computational Chemistry
Abstract: The emergence of pandemic situations originated from severe acute respiratory syndrome (SARS)-CoV-2 and its new variants created worldwide medical emergencies. Due to the non-availability of efficient drugs and vaccines at these emergency hours, repurposing existing drugs can effectively treat patients critically infected by SARS-CoV-2. Finding a suitable repurposing drug with inhibitory efficacy to a host-protein is challenging. A detailed mechanistic understanding of the kinetics, (dis)association pathways, key protein residues facilitating the entry–exit of the drugs with targets are fundamental in selecting these repurposed drugs. Keeping this target as the goal of the paper, the potential repurposing drugs, Nafamostat, Camostat, Silmitasertib, Valproic acid, and Zotatifin with host-proteins HDAC2, CSK22, eIF4E2 are studied to elucidate energetics, kinetics, and dissociation pathways. From an ensemble of independent simulations, we observed the presence of single or multiple dissociation pathways with varying host-proteins-drug systems and quantitatively estimated the probability of unbinding through these specific pathways. We also explored the crucial gateway residues facilitating these dissociation mechanisms. Interestingly, the residues we obtained for HDAC2 and CSK22 are also involved in the catalytic activity. Our results demonstrate how these potential drugs interact with the host machinery and the specific target residues, showing involvement in the mechanism. Most of these drugs are in the preclinical phase, and some are already being used to treat severe COVID-19 patients. Hence, the mechanistic insight presented in this study is envisaged to support further findings of clinical studies and eventually develop efficient inhibitors to treat SARS-CoV-2
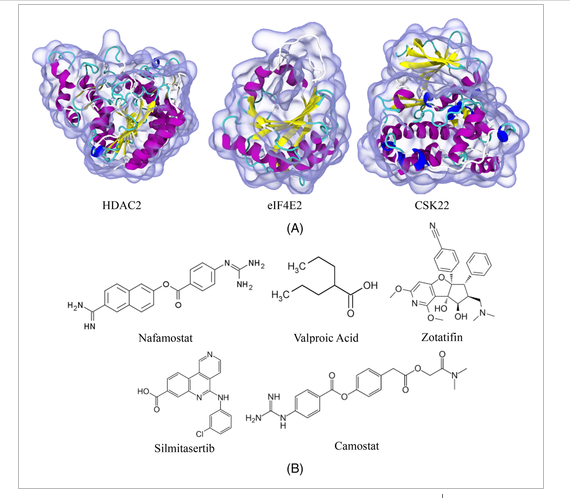
Read More ...
|
-
Parey, V, Abraham, B, Jyothirmai, and Singh JK “Mechanistic Insights for Electrochemical Reduction of CO2 into Hydrocarbon Fuels over O-Terminated MXenes” Catal. Sci. Technol.
Abstract: Two-dimensional (2D) transition metal carbides/nitrides (MXenes) have attracted intensive attention for the electrochemical reduction of CO2 into renewable fuels and chemical feedstock. Although encouraging progress has been made so far, many advances are still needed to understand and clarify the CO2RR mechanism over functionalized MXenes. In this regard, we present the promising selective conversion capabilities of group IV (Ti2X and Zr2X; X = C, N or B) MXenes with O-termination for catalyzing the carbon dioxide reduction reaction (CO2RR) to methane (CH4). The unique CO2RR behavior observed on O-terminated MXenes is mainly due to the choice of *HCOOH pathway, where the majority of electrocatalytic CO2 reductions prefer the ubiquitous *CO intermediates. The proposed reliable scaling relationships demonstrate better coordination between the reaction intermediates and binding energies of *COOH/*HCOOH, thereby indicating its importance as a key descriptor to assess the catalytic performance of O-terminated MXenes. The catalytic selectivity for the CO2RR is higher than HER, indicating that the selectivity between them is crucial and depends on the efficiency of the catalyst. Our theoretical findings provide useful insight and opportunities to develop advanced MXene based catalysts for electrochemical CO2 reduction into valuable chemicals and fuels.
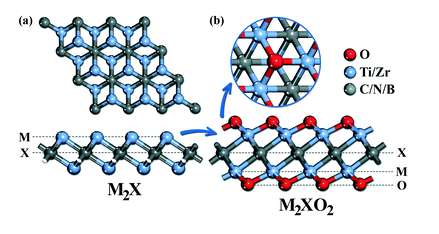
Read More ...
|
-
Abraham M, Parey V, Jyothirmal MV, Singh JK “Tuning the structural properties and chemical activities of graphene and hexagonal boron nitride for efficient adsorption of steroidal pollutants” Applied Surface Science
Abstract: Two-dimensional (2D) materials are in general considered as incredible recognition functions toward different targets. Here, our aim is to understand whether the target steroidal pollutants can adsorb on 2D surfaces or not? For this purpose, we have performed first-principles calculations to analyze the molecular interactions involved in the adsorption mechanism of target steroidal estrogens (estrone, E1; 17-estradiol, E2 and estriol, E3) and anti-estrogens (bisphenol-A, BPA) on pure and defective graphene and boron nitride (h-BN) surfaces. After screening 144 configurations of both pure and defective graphene and h-BN surfaces, we found that the nitrogen vacancy in h-BN (VBN) surface is highly sensitive and selective towards target steroid molecules with apparent charge transfer and robust adsorption energy. The contrasted chemical reactivity of h-BN and graphene demonstrates that the target steroid pollutants are chemisorbed over VBN, while this mechanism is not spotted on pure and defective graphene surface. The decreased distance between VBN and steroid molecules is due to electronic rearrangement, leading to an attraction of B-atoms of VBN toward target steroid molecules. Marked variations are also observed on the electronic structure of VBN after the adsorption of target steroids, which indicates significant charge transfer from steroid molecules to corresponding surfaces. The observed conspicuous variation between the two systems can be related to their electronic properties and the availability of insufficient screening patterns for graphene surface when compared to that of h-BN. These fundamental findings may furnish novel insights into the rational design and development of defect engineered h-BN for the detection of critically relevant steroid pollutants.
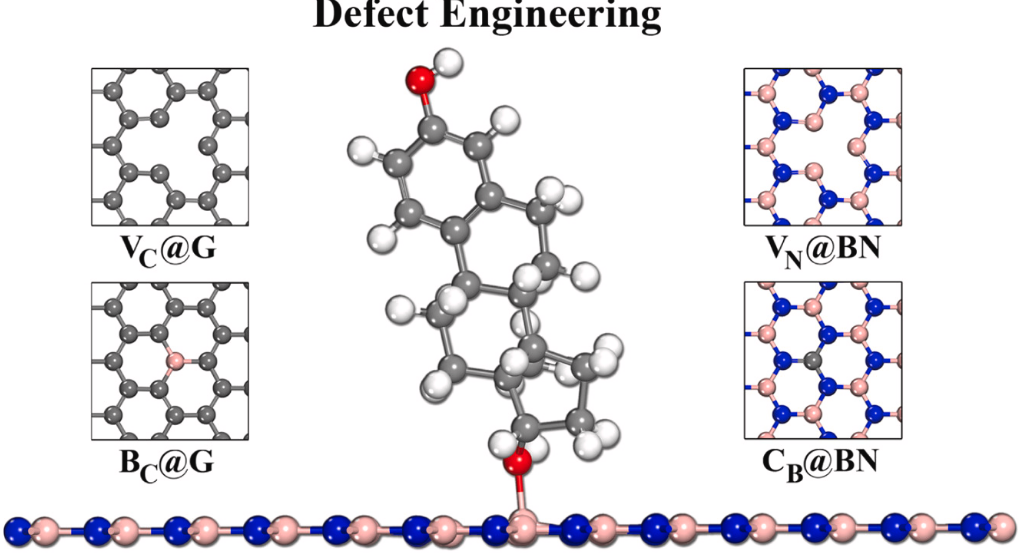
Read More ...
|
|
|
|
2021
|
|---|
|
|
|
|
|
|
|
|
-
Parey V, Abraham BM, Mir S and Singh JK “High-throughput screening of atomic defects in MXenes for CO2 capture, activation and dissociation” ACS Applied Materials and Interfaces
Abstract: The capture, activation, and dissociation of carbon dioxide (CO2) is of fundamental interest to overcome the ramifications of the greenhouse effect. In this regard, high-throughput screening of two-dimensional MXenes has been examined using well-resolved first-principles simulations through DFT-D3 dispersion correction. We systematically investigated different types of structural defects to understand their influence on the performance of M2X-type MXenes. Defect calculations demonstrate that the formation of M2C(VMC) and M2N(VMN) vacancies require higher energy, while M2C(VC) and M2N(VN) vacancies are favorable to form during the synthesis of M2X-type MXenes. The M2X-type MXenes from group III to VII series show remarkable behavior for active capturing of CO2, especially group IV (Ti2X and Zr2X) MXenes exhibit unprecedentedly high adsorption energies and charge transfer (>2e) from M2X to CO2. The potential CO2 capture, activation, and dissociation abilities of MXenes are emanated from Dewar interactions involving hybridization between pi orbitals of CO2 and metal d-orbitals. Our high-throughput screening demonstrates chemisorption of CO2 on pure and defective MXenes, followed by dissociation into CO and O species.
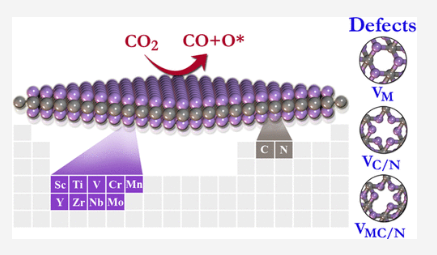
Read More ...
|
-
Kumawat A, Namsani S,Pramanik D and Singh JK “Integrated docking and enhanced sampling based selection of repurposing drugs for SARS-CoV-2 by targeting host dependent factors” Journal of Biomolecular Structure and Dynamics}
Abstract: Since the onset of global pandemic, the most focused research currently in progress is the development of potential drug candidates and clinical trials of existing FDA approved drugs for other relevant diseases, in order to repurpose them for the COVID-19. At the same time, several high throughput screenings of drugs have been reported to inhibit the viral components during the early course of infection but with little proven efficacies. Here, we investigate the drug repurposing strategies to counteract the coronavirus infection which involves several potential targetable host proteins involved in viral replication and disease progression. We report the high throughput analysis of literature-derived repurposing drug candidates that can be used to target the genetic regulators known to interact with viral proteins based on experimental and interactome studies. In this work we have performed integrated molecular docking followed by molecular dynamics (MD) simulations and free energy calculations through an expedite in silico process where the number of screened candidates reduces sequentially at every step based on physicochemical interactions. We elucidate that in addition to the pre-clinical and FDA approved drugs that targets specific regulatory proteins, a range of chemical compounds (Nafamostat, Chloramphenicol, Ponatinib) binds to the other gene transcription and translation regulatory proteins with higher affinity and may harbour potential for therapeutic uses. There is a rapid growing interest in the development of combination therapy for COVID-19 to target multiple enzymes/pathways. Our in silico approach would be useful in generating leads for experimental screening for rapid drug repurposing against SARS-CoV-2 interacting host proteins.
Read More ...
|
|
|
|
|
|
|
-
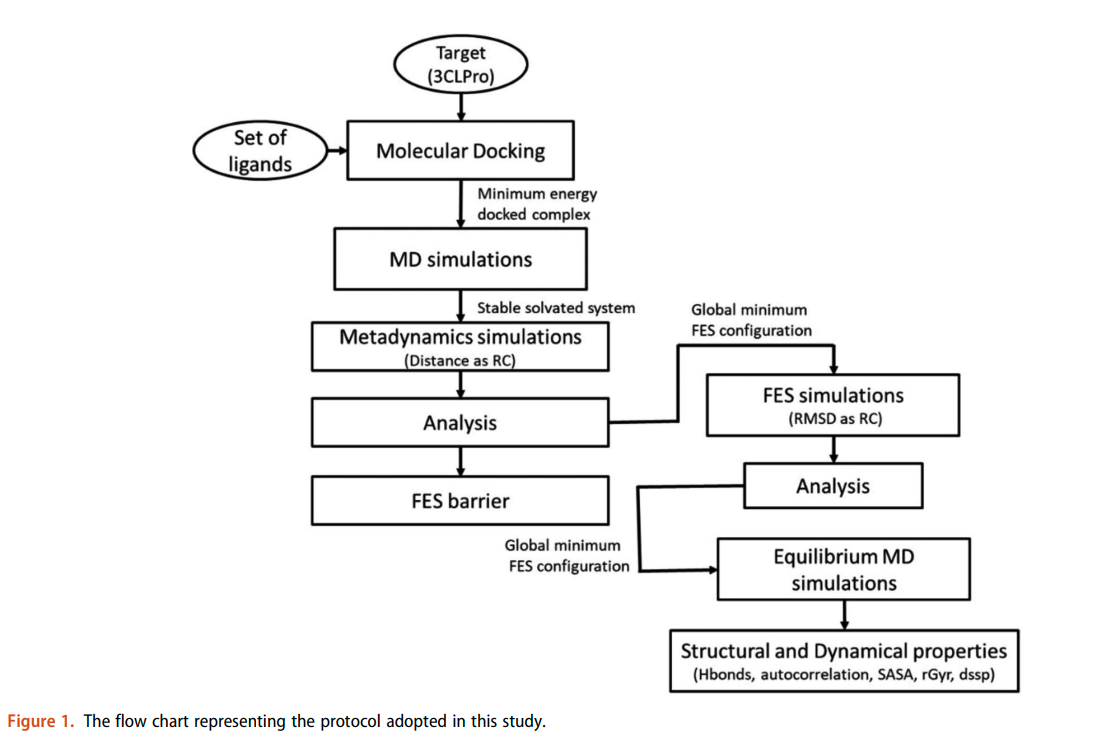
Namsani S, Pramanik D, Khan MA, Roy S and Singh JK “Potential drug candidates for SARS-CoV-2 using computational screening and enhanced sampling methods” Bio. Medi. Chem.
Abstract: Here, we report new chemical entities that exhibit highly specific binding to the 3 chymotrypsin like cysteine protease present in the novel severe acute respiratory syndrome coronavirus 2 . Because the viral 3 CLpro protein controls coronavirus replication, 3 CLpro is identified as a target for drug molecules. We implemented an enhanced sampling method in combination with molecular dynamics and docking to reduce the computational screening search space to four molecules that could be synthesized and tested against. Our computational method is much more robust than any other method available for drug screening because of sampling of the free energy surface of the binding site of the protein and use of explicit solvent. We have considered all possible interactions between all the atoms present in the protein, ligands, and water. Using high performance computing with graphical processing units, we were able to perform a large number of simulations within a month and converge the results to the four most strongly bound ligands from a set of 17 ligands with lower docking scores. Additionally, we have considered N3 and 13b ketoamide inhibitors as controls for which experimental crystal structures are available. Out of the top four ligands, PI 06 was found to have a higher screening score compared to the controls. Based on our results and analysis, we confidently claim that we have identified four potential ligands, out of which one ligand is the best choice based on free energy and the most promising candidate for further synthesis and testing against.
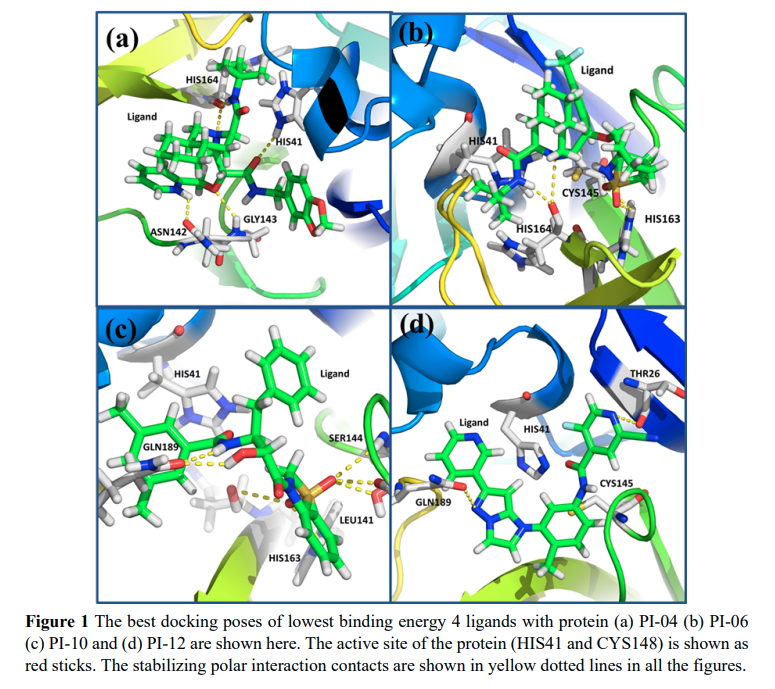
Read More ...
|
|
|
|
2020
|
|---|
-
Saha P,Yadav V, Gurunarayanan V, Ramapanicker R, Singh JK, Gopakumar T “Revealing the Limits of Intermolecular Interactions: Molecular Rings of Ferrocene Derivatives on Graphite Surface” J. Phys. Chem. Lett.
Abstract: We report the formation of discrete molecular rings/spirals of small molecules (1,3-dithia derivatives of ferrocene) on a highly oriented pyrolytic graphite (HOPG) surface. On the basis of microscopy and theoretical calculations, molecular level arrangement within the molecular rings is understood. The molecular rings show a limiting inner diameter, and we interpret it to be related to the critical intermolecular interaction limit. This limiting value of the inner diameter is surprisingly correlated with that observed for molecular rings/disks of a few reported molecules. The correlation reveals that molecular rings formed typically by weak van der Waals interactions should always show a limiting inner diameter and should be independent of molecular structure, size, and chemical nature.
Read More ...
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
-
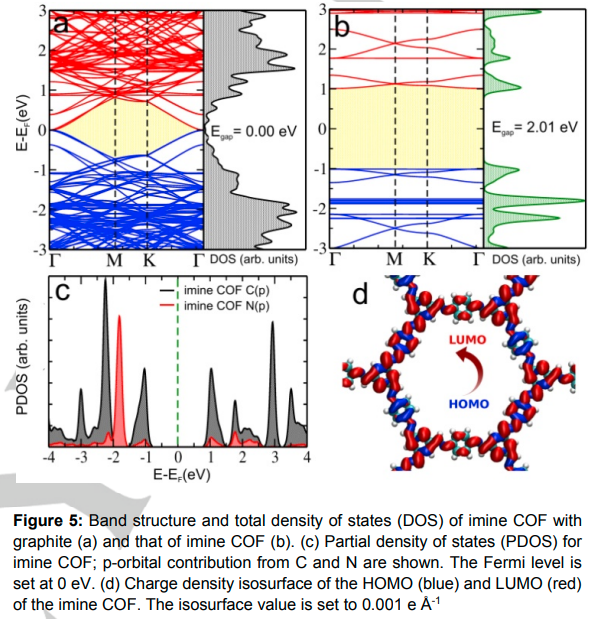
Yadav VK, Mir S, Mishra V, Gopakumar TG and Singh JK “A Simple Molecular Design for Tunable Two-Dimensional Imine Covalent Organic Frameworks for Optoelectronic Applications” Phys. Chem. Chem. Phys.
Abstract: Two-dimensional covalent organic frameworks (2D-COFs) belong to a new class of molecular materials that have attracted huge attention in recent years due to their analogous nature to graphene. In this work, we present a systematic study of the electronic structure, carrier mobility and work function of imine based 2D-COFs. We identify these 2D-COFs as a new class of semiconducting materials with tunable electronic/optoelectronic properties and significant mobility. The results show that by rationally doping 2D-COFs at the molecular level, it is possible to control their structural and optoelectronic responses. Cohesive energy calculations revealed that all the studied 2D-COFs are thermodynamically stable. Also, the calculated binding energy of 2D-COFs on HOPG was found to be less than 1 eV, which indicates that the COFs do not interact strongly with HOPG, and it will not affect their electronic properties. Additionally, we have synthesized a 2,4,6-pyrimidinetriamine based 2D-COF and experimentally measured its band gap using scanning tunnelling spectroscopy. The experimentally measured band gap is found to be in good agreement with theoretical results.
Read More ...
|
|
|
|
|
|
|
|
2019
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
-
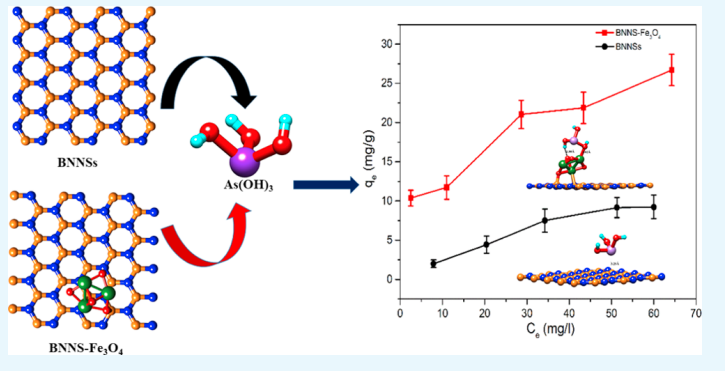
Bhangari RS, Singh AK, Namsani S, Singh JK and Sinha N “Magnetite-Coated Boron Nitride Nanosheets for the Removal of Arsenic (V) from Water” ACS Appl. Mater. Interfaces
Abstract: It is widely known that the existence of arsenic (As) in water negatively affects humans and the environment. We report the synthesis, characterization, and application of boron nitride nanosheets (BNNSs) and Fe3O4-functionalized BNNS (BNNS–Fe3O4) nanocomposite for removal of As(V) ions from aqueous systems. The morphology, surface properties, and compositions of synthesized nanomaterials were examined using scanning electron microscopy, transmission electron microscopy, X-ray powder diffraction, surface area analysis, zero-point charge, and magnetic moment determination. The BNNS–Fe3O4 nanocomposites have a specific surface area of 119 m2 g–1 and a high saturation magnetization of 49.19 emu g–1. Due to this strong magnetic property at room temperature, BNNS–Fe3O4 can be easily separated in solution by applying an external magnetic field. From the activation energies, it was found that the adsorption of As(V) ions on BNNSs and BNNS–Fe3O4 was due to physical and chemical adsorption, respectively. The maximum adsorption capacity of BNNS–Fe3O4 nanocomposite for As(V) ions has been found to be 26.3 mg g–1, which is 5 times higher than that of unmodified BNNSs (5.3 mg g–1). This closely matches density functional theory simulations, where it is found that binding energies between BNNS–Fe3O4 nanocomposite and As(OH)5 are 5 times higher than those between BNNSs and As(OH)5, implying 5 times higher adsorption capacity of BNNS–Fe3O4 nanocomposite than unmodified BNNSs. More importantly, it was observed that the synthesized BNNS–Fe3O4 nanocomposite could reduce As(V) ion concentration from 856 ppb in a solution to below 10 ppb (>98.83% removal), which is the permissible limit according to World Health Organization recommendations. Finally, the synthesized adsorbent showed both separation and regeneration properties. These findings demonstrate the potential of BNNS–Fe3O4 nanocomposite for commercial application in separation of As(V) ions from potable and waste water streams.
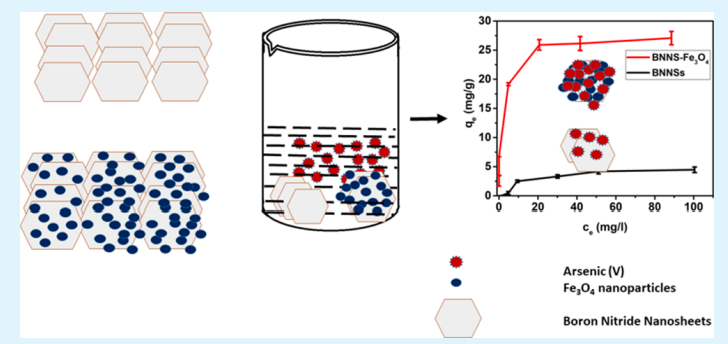
Read More ...
|
-
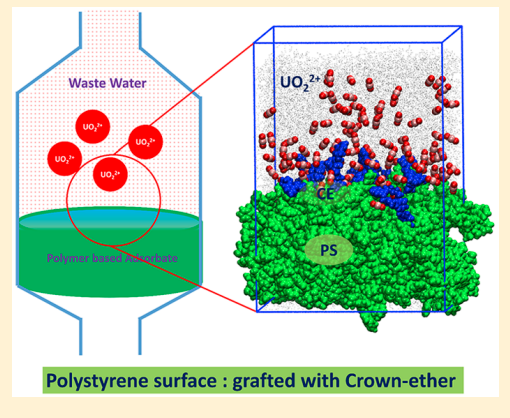
Sappidi P, Boda A, Ali SK and Singh JK “Adsorption of gadolinium (Gd3+) ions on the di benzo crown ether (DBCE) and di cyclo hexano crown ether (DCHCE) grafted on the polystyrene surface: Insights from all-atom molecular dynamics simulations and experiments” J. Phys. Chem. C
Abstract: All atom molecular dynamics simulations and experiments were performed to understand the adsorption behavior of gadolinium ion on the crown ethers grafted polystyrene surface. Two different types of crown ethers, viz, dibenzo crown ether and dicyclo hexano crown ether, were grafted separately on the PS surface to understand the adsorption behavior. We investigate the roles of ion concentration and grafting density of the crown ether on the adsorption behavior of ion on the PS surface. The adsorption of shows an increasing trend with increasing salt concentration, for all cases of crown ether grafting densities. The adsorption behavior follows the Langmuir isotherm model. We further investigate the dynamics of the ion by evaluating the diffusion coefficient and mean residence time. It was found that D decreases with increasing salt concentration for both DBCE and DCHCE. On the contrary, as expected, the value of increases with an increase in salt concentration. Overall, a 3-fold increase in was seen with increasing salt concentration. The potential of mean force analysis using umbrella sampling reveals favorable binding energy for higher grafting density of DCHCE compared to that of DBCE
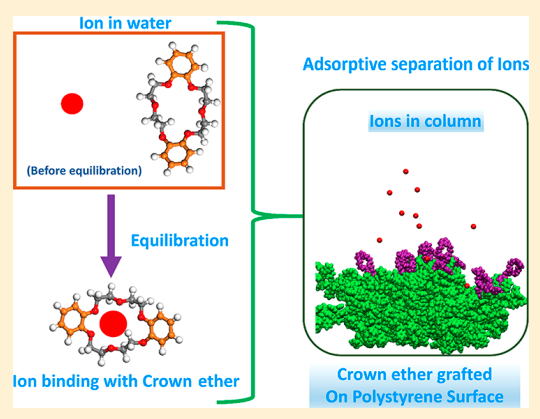
Read More ...
|
|
|
-
Saha P,Yadav V, Gurunarayanan V, Ramapanicker R, Singh JK, Gopakumar T “Understanding the Adsorption Energetics of Growth Polymorphs of Ferrocene Derivatives: Microscopic Thermal Desorption Analysis” J. Phys. Chem. C
Abstract: The ultrathin films of 2-ferrocenyl-1,3-dithiolane (FcS2C3) and 2-ferrocenyl-1,3-dithiane (FcS2C4) drop-casted from toluene on highly oriented pyrolytic graphite (HOPG) surface are investigated using atomic force microscopy (AFM). Two types of growth polymorphs have been observed, which are distinctly different based on their nature of growth and the molecular level packing. We have developed a new type of temperature-dependent desorption experiment named “microscopic thermal desorption analysis” (MTDA) for understanding the adsorption energetics related to the observed growth polymorphs on the surface. Using MTDA, we have calculated the adsorption energies of growth polymorphs of both molecules and further revealed that their formation requires an activation energy. The subtle relation between the adsorption energies and activation energies of growth polymorphs account for their average abundance on the surface. The experimental observations are further supported by density functional theory (DFT) calculations
Read More ...
|
|
|
-
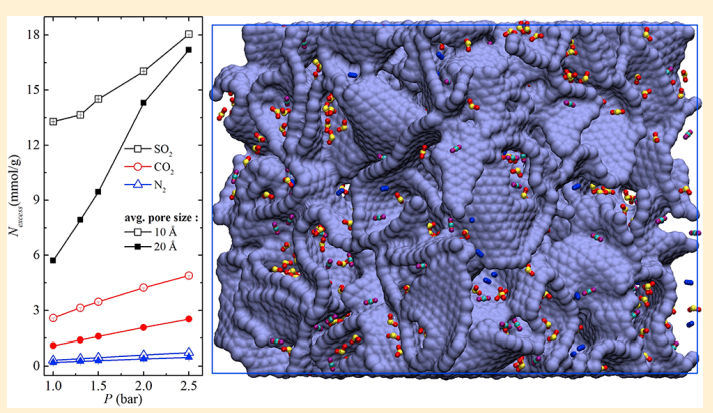
X Peng, JM Vicent-Luna, SK Jain, Q Jin and JK Singh “Computational Study of the Effect of Functional Groups on Water Adsorption in Mesoporous Carbons: Implications for Gas Adsorption” ACS Appl. Nano Materials
Abstract: The adsorption and diffusion of water in realistic carbon models at 300 K has been studied via grand canonical Monte Carlo and molecular dynamics simulations. The presence of functional groups has been found to be crucial to describe the adsorption process, while the models without functional groups are unable to capture the host–guest interactions. Functional groups were attached in cylindrical shells on the outer walls of CMK models in 2 and 4 cylindrical shells to study the effect of their concentration and location on water adsorption. The adsorption isotherm starts at a lower chemical potential, with water adsorbed near functional groups forming small clusters. On increasing chemical potential the water cluster grows and merges to form bigger clusters and completely fills the pores. We also analyzed the isosteric heat, radial distribution functions, hydrogen bond, and cluster size of water molecules. It indicates that the adsorption occurs due to the formation and growth of water clusters. For models with functional groups, the pore filling happens at lower chemical potential, when compared to the models without functional groups, owing to the presence of active sites which favors the nucleation of water molecules. The effect of functional groups is also remarkable in the diffusion of water inside the pores of models, lowering the mobility of the adsorbed molecules. The agreement between the results of the models with functional groups and the experimental observations makes the presence of these groups necessary to study the adsorption and diffusion of water in carbon models.
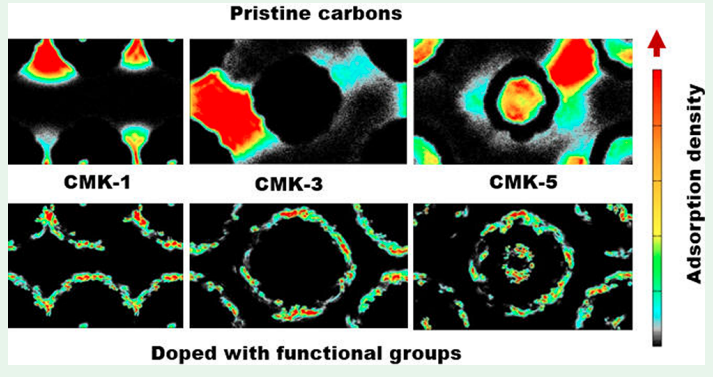
Read More ...
|
|
|
|
|
|
2018
|
|---|
-
Sappidi P, Namsani S, Ali S and Singh JK “Extraction of Gd3+ and UO22+ Ions Using Polystyrene Grafted Dibenzo Crown Ether (DB18C6) With Octanol and Nitrobenzene: A Molecular Dynamics Study” J. Phys. Chem. B
Abstract: Atomistic molecular dynamics simulations are performed in order to derive thermodynamic properties important to understand the extraction of gadolinium and uranium dioxide with dibenzo crown ether in nitrobenzene and octanol solvents. The effect of polystyrene graft length, on DBCE, on the binding behavior of is investigated for the first time. Our simulation results demonstrate that the binding of onto the oxygens of crown ethers is favorable for polystyrene grafted crown ether in the organic solvents OCT and NB. The metal ion binding free energy in different solvent environments is calculated using the thermodynamic integration method. GBinding becomes more favorable in both solvents, NB and OCT, with an increase in the polystyrene monomer length. The metal ion transferability from an aqueous phase to an organic phase is estimated by calculating transfer free-energy calculations . GTransfer is significantly favorable for both Gd3+ and UO22+ for the transfer from the aqueous phase to the organic phase (i.e., NB and OCT) via ion-complexation to DBCE with an increase in polystyrene length. The partition coefficient (log P) values for Gd3+ and UO22+ show a 5-fold increase in separation capacity with polystyrene grafted DBCE. We corroborate the observed behavior by further analyzing the structural and dynamical properties of the ions in different phases.
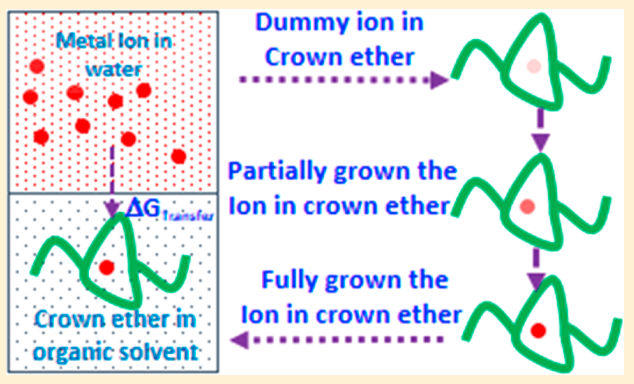
Read More ...
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2017
|
|---|
|
|
-
Bhandary D, Velachi V, Bhandary D, Dias Soeiro Cordeiro MN and Singh JK “Janus Gold Nanoparticles from Nano Droplets of Alkyl Thiolates: A Molecular Dynamics Study” Langmuir
Abstract: Janus particles provide an asymmetry in structure, which can impart diverse functionalities leading to immense importance in various applications, ranging from targeted delivery to interfacial phenomena, including catalysis, electronics, and optics. In this work, we present results of a molecular dynamics study of the growth mechanism of coating on gold nanoparticles from droplets of n-alkyl thiols with different chain lengths and terminal groups. The effect of chain lengths and functional groups on the formation of a monolayer of alkyl thiols on AuNPs is investigated. A two-step mechanism, initiated by the binding of the droplet to the nanoparticle surface with a time constant on the order of 1 ns, followed by the diffusion-driven growth with a larger time constant, is shown to capture the growth dynamics of the monolayer. It is observed that the time required for complete wetting increases with an increase in the chain length. Moreover, the monolayer formation is slowed down in the presence of carboxyl groups because of strong hydrogen bonding. The kinetics of the n-alkyl thiols coating on the nanoparticles is found to be independent of the droplet size but carboxyl-terminated thiols spread more with increasing droplet size. Furthermore, different time constants for different chains and functional groups yield Janus coating when two droplets of alkyl thiols with different terminal groups are allowed to form monolayers on the nanoparticle. The Janus balance for different combinations of alkyl thiols and nanoparticle sizes varies in the range of 0.42–0.71.
Read More ...
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2016
|
|---|
|
|
-
Rahimi M, Singh JK and Muller-Plathe F “Adsorption and Separation of Binary Mixtures of SO2, CO2 and N2 by Ordered Carbon Nanotube Arrays: Grand-canonical Monte Carlo Simulations” Phys. Chem. Chem. Phys.
Abstract: The adsorption and separation behavior of SO2–CO2, SO2–N2 and CO2–N2 binary mixtures in bundles of aligned double-walled carbon nanotubes is investigated using the grand-canonical Monte Carlo (GCMC) method and ideal adsorbed solution theory. Simulations were performed at 303 K with nanotubes of 3 nm inner diameter and various intertube distances. The results showed that the packing with an intertube distance d = 0 has the highest selectivity for SO2–N2 and CO2–N2 binary mixtures. For the SO2–CO2 case, the optimum intertube distance for having the maximum selectivity depends on the applied pressure, so that at p < 0.8 bar d = 0 shows the highest selectivity and at 0.8 bar < p < 2.5 bar, the highest selectivity belongs to d = 0.5 nm. Ideal adsorbed solution theory cannot predict the adsorption of the binary systems containing SO2, especially when d = 0. As the intertube distance is increased, the ideal adsorbed solution theory based predictions become closer to those of GCMC simulations. Only in the case of CO2–N2, ideal adsorbed solution theory is everywhere in good agreement with simulations. In a ternary mixture of all three gases, the behavior of SO2 and CO2 remains similar to that in a SO2–CO2 binary mixture because of the weak interaction between N2 molecules and CNTs.
Read More ...
|
-
Yang Y, Rahimi M, Singh JK,Bohem M and Muller-Plathe F “Adsorption and Condensation of SO2 in Double-walled Carbon Nanotube Arrays Studied by Monte Carlo Simulations and Simple Analytical Models” J. Phys. Chem. C
Abstract: Carbon nanotubes (CNTs) have been identified as extremely promising candidates for gas capture and storage. Therefore, an understanding of the adsorption mechanisms is crucial to the improvement of CNT applications. In this work, grand-canonical Monte Carlo simulations and analytical models are used to study, at the temperature of T = 303 K, the adsorption and condensation of SO2 in hexagonal arrays of double-walled CNTs of different inner nanotube radii Rin and intertube distances d. For both the inner and the outer adsorption, type I and type IV adsorption isotherms (IUPAC classification) are observed; they can be described adequately by analytical models. At a given pressure, the maximum adsorption among different CNT geometries depends strongly on the applied pressure. For the inner adsorption, three stages of adsorption are identified with increasing pressures. At low pressures, only one monolayer is formed, where the adsorption energy dominates the adsorption. At intermediate and high pressures, multilayers are formed until finally condensation is achieved; now it is the surface area or the available volume per CNT mass unit that dominates the adsorption. The nonlinear dependence of the outer adsorption on Rin and d can be explained by similar arguments as adopted for the inner adsorption. The effective number density of SO2 molecules and isosteric heat of adsorption are also analyzed to deepen our understanding of the adsorption behavior.
Read More ...
|
|
|
-
Katiyar P,Patra TK, Sarkar D, Pramik A and Singh JK “Understanding adsorption behavior of silica nanoparticles over a cellulose surface in an aqueous medium” Chem. Eng. Sci.
Abstract: The suspension and adsorption of silica nanoparticles on a cellulose surface, in an aqueous medium is investigated using Brownian dynamic simulations. The inter particle and particle–surface interactions are modeled within the framework of the DLVO theory. Our analysis predicts the accumulation of negatively charged nanoparticles near a negatively charged surface depending on the Debye screening length of the medium. A crossover from the suspension to the adsorption of negatively charged silica nanoparticles onto a negatively charged cellulose surface has been reported as the screening length () of the medium increases. The crossover is observed at , due to the interplay between the nanoparticle–nanoparticle and the nanoparticle–surface interactions. The adsorption behavior of nanoparticles is explained using the potential of mean force analysis. The amount of nanoparticles adsorbed depends on their bulk volume fraction and the screening length of the medium. Further, the effects of electrical potentials of nanoparticle and surface on the adsorption are reported. The data suggests that the adsorption of nanoparticles increases either with increasing magnitude, or/and, with decreasing magnitude. The adsorbed particles form a disordered monolayer, and undergo subdiffusive motion. We have also observed a transition from the gas-like structure to the liquid-like structure of nanoparticles in the adsorbed monolayer as their bulk volume fraction increases.
Read More ...
|
|
|
-
Halder P,Maurya M, Jain SK and Singh JK “Understanding Adsorption of CO2, N2, CH4 and its Mixture in Functionalized Carbon Nanopipe Arrays” Phys. Chem. Chem. Phys.
Abstract: The selective adsorption behaviours of carbon dioxide, methane and nitrogen on bundles of functionalized CMK-5 are investigated at 303 K using grand-canonical Monte Carlo simulations. Functional groups cause a significant enhancement in CO2 uptake . On the other hand, the adsorption amount of methane decreases with respect to bare CMK-5 by 13% (at 38.13 bar) upon functionalization. Furthermore, functionalized CMK-5 with different pore sizes (4 nm, 6 nm, 8 nm) and inter-tube distances (d = 0 to 1.5 nm) are used to investigate the adsorption behaviour of flue gases.
Read More ...
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2015
|
|---|
|
|
|
|
|
|
-
Sadanandam N, Nisanth NN and Singh JK “Interaction Potential Models for Bulk ZnS, ZnS Nanoparticle, and ZnS Nanoparticle-PMMA From First-Principles” J. Comput. Chem.
Abstract: An ab initio derived transferable polarizable force-field has been developed for Zinc sulphide (ZnS) nanoparticle (NP) and ZnS NP-PMMA nanocomposite. The structure and elastic constants of bulk ZnS using the new force-field are within a few percent of experimental observables. The new force-field show remarkable ability to reproduce structures and nucleation energies of nanoclusters (Zn1S1-Zn12S12) as validated with that of the density functional theory calculations. A qualitative agreement of the radial distribution functions of Zn-O, in a ZnS nanocluster-PMMA system, obtained using molecular mechanics molecular dynamics (MD) and ab initio MD (AIMD) simulations indicates that the ZnS–PMMA interaction through Zn-O bonding is explained satisfactorily by our force-field. 2015 Wiley Periodicals, Inc.
Read More ...
|
|
|
|
|
|
|
-
Rahimi M, Babu, D, Singh JK, Yang Y, Schneider JJ and Muller-Plate F “Double-Walled Carbon Nanotube Array for CO2 and SO2 Adsorption” J . Chem. Phys.
Abstract: Grand-canonical Monte Carlo simulations and adsorption experiments are combined to find the optimized carbon nanotube (CNT) arrays for gas adsorption at low pressures and 303 K. Bundles of 3D aligned double-walled carbon nanotube (DWCNT) with inner diameter of 8 nm and different intertube distances were made experimentally. The experimental results show that decreasing intertube distance leads to a significant enhancement in carbon-dioxide (CO2) adsorption capacity at 1 bar. The molecular simulation study on CO2 adsorption onto bundles of 3D aligned DWCNT with inner diameters of 1, 3, and 8 nm and intertube distance of 0-15 nm shows that the intertube distance plays a more important role than the CNT diameter.
Read More ...
|
|
|
|
2014
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
-
Ramarez R,Singh JK “Muller-Plate F and Bohm, M, Ice and water droplets on graphite: a comparison of quantum and classical simulations” The Journal of Chemical Physics
Abstract: Ice and water droplets on graphite have been studied by quantum path integral and classical molecular dynamics simulations. The point-charge q-TIP4P/F potential was used to model the interaction between flexible water molecules, while the water-graphite interaction was described by a Lennard-Jones potential previously used to reproduce the macroscopic contact angle of water droplets on graphite. Several energetic and structural properties of water droplets with sizes between 102 and 103 molecules were analyzed in a temperature interval of 50–350 K. The vibrational density of states of crystalline and amorphous ice drops was correlated to the one of ice Ih to assess the influence of the droplet interface and molecular disorder on the vibrational properties. The average distance of covalent OH bonds is found 0.01 larger in the quantum limit than in the classical one. The OO distances are elongated by 0.03 in the quantum simulations at 50 K. Bond distance fluctuations are large as a consequence of the zero-point vibrations. The analysis of the H-bond network shows that the liquid droplet is more structured in the classical limit than in the quantum case. The average kinetic and potential energy of the ice and water droplets on graphite has been compared with the values of ice Ih and liquid water as a function of temperature. The droplet kinetic energy shows a temperature dependence similar to the one of liquid water, without apparent discontinuity at temperatures where the droplet is solid. However, the droplet potential energy becomes significantly larger than the one of ice or water at the same temperature. In the quantum limit, the ice droplet is more expanded than in a classical description. Liquid droplets display identical density profiles and liquid-vapor interfaces in the quantum and classical limits. The value of the contact angle is not influenced by quantum effects. Contact angles of droplets decrease as the size of the water droplet increases which implies a positive sign of the line tension of the droplet
Read More ...
|
|
2013
|
|---|
|
|
-
Sharma AK, Khare P, Singh JK and Verma N “Preparation of novel carbon microfiber/carbon nanofiber-dispersed polyvinyl alcohol-based nanocomposite material for lithium-ion electrolyte battery separator” Materials Science and Engineering: C
Abstract: Abstract
A novel nanocomposite polyvinyl alcohol precursor-based material dispersed with the web of carbon microfibers and carbon nanofibers is developed as lithium (Li)-ion electrolyte battery separator. The primary synthesis steps of the separator material consist of esterification of polyvinyl acetate to produce polyvinyl alcohol gel, ball-milling of the surfactant dispersed carbon micro-nanofibers, mixing of the milled micron size (500 nm) fibers to the reactant mixture at the incipience of the polyvinyl alcohol gel formation, and the mixing of hydrophobic reagents along with polyethylene glycol as a plasticizer, to produce a thin film of 25 mu. The produced film, uniformly dispersed with carbon micro-nanofibers, has dramatically improved performance as a battery separator, with the ion conductivity of the electrolytes (LiPF6) saturated film measured as 0.119 S cm 1, approximately two orders of magnitude higher than that of polyvinyl alcohol. The other primary characteristics of the produced film, such as tensile strength, contact angle, and thermal stability, are also found to be superior to the materials made of other precursors, including polypropylene and polyethylene, discussed in the literature. The method of producing the films in this study is novel, simple, environmentally benign, and economically viable.
Read More ...
|
|
|
|
|
-
Rahimi M, Singh JK*, Babu DJ, Schneider JJ and Muller-Plathe F “Understanding carbon dioxide adsorption in carbon nanotube arrays: Molecular simulation and adsorption measurements” The Journal of Physical Chemistry
Abstract: Grand canonical Monte Carlo simulations and adsorption experiments are conducted to understand the adsorption of CO2 onto bundles of 3D aligned double walled carbon nanotubes of diameter 5 nm at 303 K. The simulation of partial adsorption isotherms, only inner tube volume, only interstices between tubes, and unrestricted, allows a breakdown of the experimental adsorption isotherms into contributions of different regions. The results are compatible with microscopic observations of the majority of the inner tube volumes being accessible for CO2. Further, the unrestricted adsorption isotherm is quantitatively equivalent to the sum of inner and outer adsorption for the pressure range considered in this work, p 40 bar, indicating no significant interference between inner and outer regions. The intertube distance, which is varied from 0 to 15 nm, dramatically affects the isosteric heat of adsorption and adsorption capacity. Excess adsorption is found to display a nonlinear behavior with d, for unrestricted and outer cases. For low pressures p 14 bar, maximum adsorption occurs at d 0.5 nm. However, for higher pressures, 14 40 bar, the adsorption peaks at d 1 nm. The Freundlich isotherm is found to fit the experimental and simulation data. The adsorption sequence changes with the intertube distance for the unrestricted case. At d 0.5 nm, adsorption proceeds with increasing loading in the following order grooves and inner surface adsorption fill interstitial region fill inner region. However, at higher distances, d 0.5 nm, the sequence changes the following inner surface adsorption partial outer surface adsorption complete outer surface adsorption fill interstitial, groove, inner adsorption. The change in mechanism of adsorption is clearly reflected in the behavior of the heat of adsorption, where we observed a crossover behavior at around d 0.5 nm.
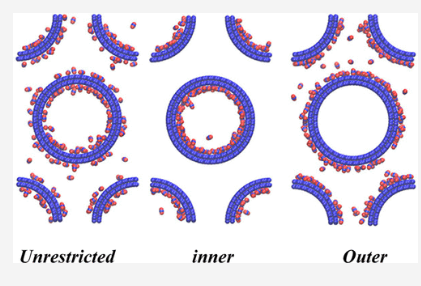
Read More ...
|
|
|
|
|
|
2012
|
|---|
|
|
|
|
|
|
|
|
-
Boda A, De S, Ali M, Tulishetti S, Khan S and Singh JK “From Microhydration to Bulk Hydration of Sr2+ Metal Ion: DFT, MP2 and Molecular Dynamics Study” Journal of Molecular Liquids
Abstract: This paper presents the results of quantum chemical and classical molecular dynamics (MD) simulations of the microhydration states of the Sr2+ ion. The quantum chemical results strongly suggest a coordination number (CN) of 8 for the first hydration shell of Sr2+, which is in quantitative agreement with data available from X-ray absorption fine structure (XAFS) measurements. The calculated theoretical Srsingle bondO bond distance of 2.59 Å is also in excellent agreement with the XAFS results (2.60 Å). Classical MD simulations are conducted on various water models to predict the hydration structure of the Sr2+ ion. The CN is found to be in the range of 8–9 using SPC, TIP3P, and TIP4P-2005 water models, with the probability more skewed toward 8. MD–EXAFS study and coordination number analyses reveal that TIP4P-2005 is the best model potential for simulating water molecules to reproduce the experimentally observed absorption spectra and coordination numbers.
Read More ...
|
|
|
|
|
|
2011
|
|---|
|
|
|
|
|
|
-
Ghosh A, Patra TK, Rishikant, Singh RK, Singh JK and Bhattacharya S “Surface Electrophoresis of ds-DNA across orthogonal pair of surfaces” App. Phys. Lett.
Abstract: A gel free microchannel device made up of polydimethyl siloxane is fabricated for the surface based electrophoresis of double stranded deoxy ribonucleic acid molecules. In the presence of directional external electric field, DNA fragments near the corners of the microchannel are found to separate faster as compared to those over the base of the channel. This is in spite of the reduction in the mobility of molecules over the channel corners. We performed coarse grained molecular dynamics simulations which reveal that, though the adsorption energy of the DNA fragments increases near the corner, it is the increase in the relative mobility which enhances the separation of the fragments over the corner.
Fractionation of ds DNA based on different molecular lengths by passing them through a sieving medium has been regularly explored for all diagnostics. Some of the widely used methods include slab gel electrophoresis,1 zone electrophoresis, 2,3 capillary electrophoresis 4 to 7 in microcapillaries, microfabricated channel arrays, etc.8,9 Fragment sizes above 10 kbp are resolved very poorly by the above bulk electrophoresis techniques.5 Thus, new schemes for DNA fractionation have emerged involving electrostatic interactions between nucleic acid molecules and surfaces using its inherent chemical, topological, or structural properties,10 to 13 as demonstrated for the first time by Pernodet et al.10 over a perfectly flat silicon wafer and later by Han and Craighead.14 Literature shows the influence of various parameters like electric field intensity, ionic strength, and migration distances on the mobility of ds DNA as they are transported along surfaces.15 Lee and Kuo16 fabricated a gel-free microchannel electrophoresis device for fractionating larger DNA fragments where they chemically modified the channel’s bottom surface. The process although widely explored has not been fully understood.
In this letter, we have investigated the surface electrophoresis process for ds DNA in polydimethyl siloxane microchannels. A peculiar behavior of ds DNA is observed as they move along the channel corners instead of the channel base in a few trials. The channel length and time needed for fractionating a 1 kb ladder decreased substantially along the channel corners as compared to the base. In other words, orthogonally placed pair of surfaces is found to be more favorable for the fractionation process in comparison to flat surfaces. We have utilized molecular dynamics simulations to understand the physics of interaction of the ds DNA with the surface to explain the above behavior.
The microchannel electrophoresis device for our experiments was fabricated in PDMS by using a carbon dioxide laser etched pattern on a poly methyl methacrylate substrate.
Read More ...
|
|
|
|
|
|
|
|
2010
|
|---|
-
Cummings PT, Docherty H, Iacovella, CR and Singh JK “Phase Transitions in nanoconfined fluid: The evidence from simulation and theory (PERSPECTIVE ARTICLE)” AICHE J.
Abstract: Nanoconfined fluids — that is, fluids confined between surfaces separated by nanometers — play important roles in many natural and man-made processes and products. One example is hard disk drive lubrication where, as data density has increased exponentially, the distance between the read head and rotating platen has been exponentially decreasing for several decades. This distance is now at 10–12 nm, and in the next generation of disk drives will be at 8 nm;1 currently, monolayers of lubricant are used to protect disk drives in abnormal situations (e.g., power loss), but in the future it is expected that they will be lubricated at all times, including during read/write operations. Additional examples include the lubrication of microelectromechanical systems (MEMS), and nanoelectromechanical systems (NEMS),2 and a model3 for the natural lubrication of synovial joints, all of which can involve moving surfaces separated by distances of the order of nm. The latter exhibit very low-sliding friction at normal pressures up to 5 MPa or more; the model system, consisting of polyzwitterionic brushes polymerized directly onto the mica sheets in a surface force balance (SFB), exhibits very similar low-sliding friction (within a factor of 2 of the natural synovial joints) at pressures as high as 7.55 MPa. These three examples highlight the desirability of being able to lubricate effectively between surfaces moving relative to each other while separated by distances on the order of nm. If the lubricant undergoes a fluid-solid phase transition under nanoconfinement, resulting in a many order of magnitude increase in the effective viscosity and the onset of a nonzero yield stress,* then it is clearly not useful as a lubricant. In addition to lubrication at the nanoscale, phase transitions under nanoconfinement are also clearly important in industrial adsorption and catalytic processes (micro- and mesoporous adsorbents, with pore widths of under 2 nm and 2–50 nm, respectively, are widely used in the chemical, petrochemical, gas processing, and pharmaceutical industries for separations, pollution abatement, and as catalysts and catalyst supports).4 Additional application areas (e.g., in geology, oil recovery and nanofabrication, including nanotemplating through nanoconfinement) are described in the excellent review article by Gelb etal.4

Read More ...
|
|
|
|
|
|
|
|
|
|
2009
|
|---|
-
Bhateja A, Singh JK, and Sharma I “Axial segregation in horizontally vibrated granular materials: A numerical study.” Advances in Geomechanics, Korean Journal of Civil Engineering.
Abstract: It is known that a horizontally vibrated binary mixture in a tapered and inclined channel segregates axially, with the two species moving to the opposite ends of the channel. In general, the parameters that affect the segregation process include the forcing frequency and its amplitude, the constituents’ mass and size, and the taper and inclination of the channel. The ultimate goal here is to locate those parameters that are most significant to the segregation process, thereby providing control variables for practical applications. However, owing to the complexity of the problem, as a first step to better understand the physics behind this phenomenon, we undertake three dimensional molecular dynamics simulations of a horizontally vibrated mono-disperse granular particles in a tapered and inclined channel. Though at this stage, the immediately addressed problem is of more relevance to the granular material industry, it is envisaged that tools developed to understand this process will ultimately have wide applicability to granular systems, occurring in both natural contexts and in geotechnical engineering.
Read More ...
|
|
|
|
|
-
Singh SK, Sinha A, Deo G, and Singh JK “Vapor-liquid phase coexistence, critical properties and surface tension of confined alkanes” J. Phys. Chem. C
Abstract: Configurational-bias grand-canonical transition-matrix Monte Carlo simulations are conducted to investigate various thermophysical properties, such as phase coexistence, critical properties, density and orientation profiles of liquid and vapor phases, and vapor liquid surface tension of methane, ethane, propane, n butane, and n octane in bulk and slit pores of graphite and mica surfaces. An exponential-6 model is used for the investigation of normal alkanes with a cutoff radius, 15 . It is found that the surface tension of the bulk n-alkane, based on a truncated exp-6 model, agrees reasonably well with the experimental data. Critical properties are reported by means of the rectilinear diameter approach and least-squares technique. The shift in the critical temperature under confinements follows more than two linear regimes with an inverse in the slit width, as the slit width approaches the two-dimensional limit. This is contrary to what has been previously reported in the literature. The behavior of the critical temperature shift is sensitive to the nature of the surface. The critical density, on the other hand, fluctuates with a decrease in the slit width. The shift in the critical vapor pressure continuously increases with a decrease in the slit width toward the two dimensional value and becomes constant for pore sizes less than 5 for the fluid studied in this work. Corresponding state plots suggest that the deviation of the saturation vapor pressure from the bulk saturation pressure under confinement is positive for large pores and negative for smaller pores. Vapor liquid surface tension values for n alkanes, in both types of slit pores, are computed via the finite size scaling method of Binder and compared with their bulk values. Our investigation reveals that under slit pore confinement the vapor liquid surface tension decreases many fold.
Read More ...
|
|
|
|
|
|
|
|
|
|
2008
|
|---|
|
|
-
Sachdeva S, Ram RP, Singh JK, and Kumar A “Synthesis of Anion Exchange Polystyrene Membranes for the Electrolysis of Sodium Chloride” AICHE J
Abstract: We have prepared a cross-linked polystyrene anion exchange composite membrane for the electrolysis of sodium chloride to produce sodium hydroxide by selective removal of chloride ions. The composite membrane is homogeneously modified by gas phase nitration, followed by amination using hydrazine hydrate, and further reaction with dichloroethane and triethylamine to introduce quaternary ammonium charges on it. We showed that the membrane is specific to the transport of chloride ions through its pores. The performance of the membrane has been evaluated in terms of current efficiency and power consumption, and the effect of various parameters like current density, initial salt concentration, and circulation rate is studied. The maximum current efficiency obtained is 96.5% and the corresponding power consumption is 0.1216 kWh/mol at 5.2 N initial salt concentration and current density of 254 A/m2. © 2008 American Institute of Chemical Engineers AIChE J, 2008
Read More ...
|
|
|
|
|
|
|
|
2007
|
|---|
|
|
|
|
|
|
|
|
|
|
|
2006
|
|---|
|
|
|
|
|
2005
|
|---|
|
|
|
2004
|
|---|
|
|
|
|
|
|
|
2003
|
|---|
|
|
|
|
|
2000
|
|---|
|
|